

PyMOL fornece diversos métodos para alinhamento estrutural de moléculas tanto por meio de sua interface simplificada quanto por meio de seu terminal de linhas comando. Para compreender como ele funciona na prática, vamos apresentar a seguir, uma série de estudos de caso utilizando-o para realizar alinhamentos de estruturas.
Estudo de caso 1: alinhamento entre duas beta-glicosidases
No exemplo a seguir, demonstraremos como utilizar a interface do PyMOL para realizar o alinhamento de duas proteínas. Para este estudo de caso, realizaremos o alinhamento da beta-glicosidase do fungo Humicola insolens (PDB ID: 4MDP) e da beta-glicosidase do cupim Neotermes koshunensis (PDB ID: 3VIK).
Inicialmente, acesse o terminal de comando da interface gráfica do PyMOL (Figura 10). A priori, utilizaremos o terminal apenas para fazer o download dos arquivos PDB que serão utilizados. Caso já os tenha em seu computador, você pode utilizar o menu File > Open para abrir manualmente os arquivos PDB.

Para fazer o download dos arquivos utilize o comando fetch seguido do código PDB que se deseja carregar. Para o estudo de caso, execute os comandos:
fetch 4mdp |
Isso irá carregar a visualização tridimensional das duas estruturas das proteínas beta-glicosidase do fungo Humicola insolens (PDB ID: 4MDP), representada na cor verde, e de Neotermes koshunensis (PDB ID: 3VIK), representada na cor azul (Figura 11).

Para alinhar as estruturas, podemos utilizar o painel à direita para selecionar uma ação de alinhamento (Figura 12). Vamos escolher uma das estruturas (no caso, selecionamos 3VIK) e clicar sobre o botão com a letra A (action) ao lado do nome da estrutura. Isso irá carregar um menu com diversas ações que podem ser aplicadas a estrutura desejada. Vamos até a opção align (alinhamento), depois em “to molecule (*/CA)” e por fim em 4mdp (pois iremos sobrepor 3vik a 4mdp).

Por padrão, o PyMOL realiza um alinhamento de sequências para determinar regiões similares e depois um refinamento é realizado usando as posições dos carbonos-alfa de cada resíduo de aminoácido. Como resultado, pode-se observar as estruturas sobrepostas (Figura 13).

PyMOL permite ainda visualizar o alinhamento de sequências na interface acima do alinhamento estrutural. Para isso, basta clicar no botão com a letra S no canto inferior direito (Figura 14).

PyMOL permite ainda ampliar a visualização de uma região específica. Para isso, selecione a região desejada com base na visualização das sequências alinhadas, ou clique sobre os aminoácidos desejados (você pode pressionar SHIFT para selecionar mais de um aminoácido). A seguir, clique com o botão direito sobre a sequência selecionada e vá em zoom (Figura 15).

O PyMOL irá gerar uma visualização mais próxima da região desejada (Figura 16). Você pode ainda usar o mesmo processo para centralizar a região desejada (no caso, clique na opção center). Ou ainda pode esconder a região clicando em hide, exibir os rótulos com nomes dos aminoácidos e átomos clicando em label, ou alterar as cores clicando em color.

Estudo de caso 2: alinhamento entre duas lisozimas
Para compreendermos melhor como funciona o algoritmo de alinhamento padrão do PyMOL, neste novo exemplo, realizaremos um alinhamento da estrutura de cristal de uma lisozima (PDB ID: 2LZM) com uma estrutura cuja diferença se trata de um aminoácido não natural p-iodo-L-fenilalanina na posição 153 (PDB ID 1T6H).
Para realizar o alinhamento das estruturas, PyMOL inicialmente utiliza programação dinâmica para realizar um alinhamento de sequências. Na prática, ele realiza um alinhamento usando a ferramenta BLAST com base na matriz de pontuação BLOSUM62 (não precisamos entrar em detalhes, pois esses são parâmetros-padrão de execução do BLAST). Podemos simular essa primeira etapa executando o alinhamento das sequências usando a ferramenta BLAST web (disponível em https://blast.ncbi.nlm.nih.gov). Observe o resultado do alinhamento:

Aqui podemos ver que ambas as sequências possuem 164 aminoácidos com uma identidade superior a 99% (163 dos 164 resíduos coincidem). Veja que o alinhamento nos dá uma tabela de correspondência entre resíduos. Por exemplo, a metionina na posição 1 (resíduo M1) de 2LZM é equivalente a metionina na posição 1 de 1T6H. O mesmo vale para N2, I3, F4, E5 e assim sucessivamente. De fato, essa regra valerá para quase todos os resíduos (exceto F153 de 2LZM). Por ter estruturas bastante similares, esse exemplo poderá facilitar a compreensão da primeira etapa da estratégia de alinhamento do PyMOL.
Agora que temos a tabela de correspondentes, vamos para a próxima etapa. A seguir, PyMOL coleta as coordenadas dos carbonos-alfa de cada um dos resíduos. Lembre-se que cada resíduo de aminoácido terá um único carbono-alfa localizado em sua cadeia principal e cada átomo desses terá três valores de coordenadas de posicionamento (aqui denominadas X, Y e Z). Na Figura 17 apresentamos uma visualização da estrutura de 2LZM (Figura 17A) e 1T6H (Figura 17D). Em Figura 17 B,E são apresentados apenas os carbonos-alfa de cada uma dessas proteínas e na Figura 17C,F uma linha interligando esses átomos foi inserida para destacar a forma da proteína.

PyMOL irá utilizar apenas as posições das coordenadas desses átomos para a realização da sobreposição. É importante levar em consideração que apesar das proteínas serem bastante parecidas, as coordenadas dos átomos são bastante diferentes. Na Figura 18 podemos ver que, quando as duas estruturas são plotadas em um mesmo espaço conformacional, isto é, quando abertas em uma mesma sessão do PyMOL, os átomos estão localizados em posições diferentes.
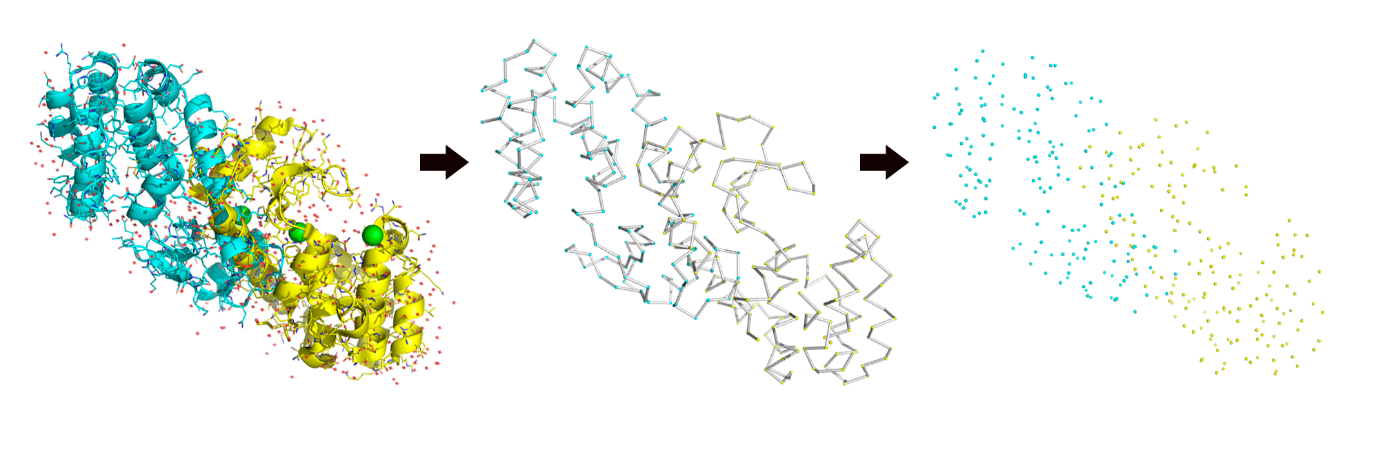
Para que possamos visualizar como uma estrutura se sobrepõe a outra, devemos alterar as coordenadas geográficas de uma das estruturas utilizando as regras de rotação e translação (lembre-se que se o algoritmo decidir rotacionar um dos átomos, todos os outros da mesma estrutura serão afetados). Assim, PyMOL utilizará a matriz de correspondência de resíduos para reposicionar os átomos da segunda proteína (no caso, escolhemos reposicionar os átomos de 1T6H).
Na Figura 19, vemos dois pares de resíduos correspondentes usados como exemplo. O primeiro deles é R125 de 2LZM, que obviamente corresponde a R125 de 1T6H. Para alinhar esses dois resíduos, a ferramenta poderia fazer um movimento de translação, movendo a posição de R125 de 1T6H para próximo da posição do resíduo equivalente em 2LZM. Perceba que ao fazer isso, todos os outros átomos seriam afetados. Para tentar melhorar esse processo, a ferramenta poderia efetuar um processo de rotação usando um segundo resíduo (no exemplo, E10). E assim, a ferramenta deveria realizar esse processo para outros resíduos, mas como ela saberia que o alinhamento está bom? Basta calcular a distância entre pontos equivalentes. Se a distância de R125 de 2LZM para R125 de 1T6H é, por exemplo, 50 Å (1 Å é equivalente a 10 10 m) e após a translação/rotação, essa distância reduziu para 1 Å, podemos concluir que o alinhamento está melhor. Após rodadas de translação e rotação, esse mesmo cálculo é feito para todos os átomos e depois é calculada uma média (chamamos isso de RMSD ou Root-mean-square deviation of atomic positions que pode ser traduzido para “desvio quadrático médio das posições atômicas”).

Na prática, o algoritmo de sobreposição é um pouco mais complicado do que o exemplo apresentado. Entretanto, o objetivo aqui é apresentar de forma didática como ele funciona de maneira geral, focando principalmente na compreensão dos movimentos de rotação e translação. Ao fim, em um alinhamento bom, as coordenadas de átomos dos carbonos-alfa irão se sobrepor e isso permitirá que possamos visualizar as proteínas sobrepostas (como visto na Figura 20).

Estudo de caso 3: alinhamento entre proteínas com sequências pouco idênticas
Nos exemplos anteriores, demonstramos a visualização de um alinhamento entre duas proteínas com estrutura conservada. Entretanto, o que aconteceria se alinhássemos duas proteínas com estruturas bastante diferentes. Para ilustrar esse hipotético problema, vamos utilizar dois PDBs escolhidos aleatoriamente: 1mdb (uma ligase) e 2lzm (uma lisozima de um bacteriófago).
Primeiro, vamos carregar os arquivos PDB de 1mdb e 2lzm usando o comando fetch:
fetch 1mdb |
E a seguir, realizar o alinhamento das duas estruturas usando a interface do PyMOL (Figura 21).

Ao tentar alinhar duas proteínas com estruturas pouco similares, PyMOL tentará encontrar a maior quantidade de regiões com sequências parecidas e tentará de alguma forma alinhá-los. Em estruturas pouco similares, isso irá gerar um alinhamento ruim. Podemos ver isso, observando as linhas amarelas que conectam pontos equivalentes em que houve a tentativa de alinhamento (Figura 22).

Observe que não há praticamente nenhuma sobreposição entre estruturas secundárias. Isso se deve ao fato do PyMOL, por padrão, usar uma estratégia baseada em alinhamento de sequências. Como as sequências apresentam uma alta discrepância (Figura 23), os poucos resíduos alinhados levarão a um alinhamento estrutural ruim.

Podemos obter resultados melhores usando os outros métodos de alinhamento presentes no PyMOL, mas para isso precisamos introduzir o uso de alinhamentos por linhas de comando.
Clique aqui para acessar a próxima seção do capítulo.
Sumário
Este texto foi retirado do capítulo “Alinhamento estrutural” de Santos e colaboradores (ainda não publicado). Para um melhor entendimento, dividimos o capítulo em sete partes:
[…] 2. Alinhamento de estruturas 3D com PyMOL […]
[…] Alinhamento de estruturas 3D com PyMOL […]