





Este manuscrito apresenta uma simulação do processo computacional para obtenção de informações estruturais e físico-químicas de proteínas, partindo apenas de uma sequência proteica.
Todas as ferramentas utilizadas neste roteiro estão disponíveis de forma online e gratuita (com a exceção do programa ChimeraX, que deve ser baixado, mas possui licença acadêmica), necessitando assim apenas de conexão com a internet para serem utilizadas.
Uma sequência modelo é apresentada ao longo do roteiro, e todas as análises são realizadas a partir dela. No entanto, os participantes são encorajados a testar e realizar o roteiro com seus próprios objetos de estudo ou interesse, além de explorar os demais parâmetros e funcionalidades disponíveis nas ferramentas que não são mencionados aqui. Em alguns momentos, também são apresentados materiais suplementares (tutoriais ou artigos de documentação) para auxiliar os alunos.
Neste artigo, abordaremos como métodos computacionais podem ser utilizados para a obtenção de informações sobre a identidade, sequência, estrutura e propriedades das proteínas.
Parte 1 – Identificação da Sequência
Para fins didáticos, utilizaremos a seguinte sequência proteica para todas as atividades subsequentes (você pode copiar e colar o conteúdo da caixa abaixo). Recomendamos que você salve a sequência em um bloco de notas, para facilitar a execução das atividades.
MLKRYLVLSVATAAFSLPSLVNAAQQNILSVHILNQQTGKPAADVTVTLEKKADNGWLQLNTAKT
DKDGRIKALWPEQTATTGDYRVVFKTGDYFKKQNLESFFPEIPVEFHINKVNEHYHVPLLLSQYGYS
TYRGS
Suponhamos que alguém te entregou a sequência acima para analisar, mas você ainda não tem nem ideia de qual é a proteína codificada por essa sequência. Como podemos então obter essa informação?
O programa BLAST [1,2] é capaz de identificar regiões de similaridade entre sequências biológicas (nucleotídeos e proteínas) fornecidas pelo usuário, compará-las com bancos de dados, e realizar o cálculo das significâncias estatísticas (neste caso, o cálculo da similaridade entre as sequências com a probabilidade de pareamento ao acaso, também chamado de E-value).
Como o nosso objetivo é identificar uma sequência proteica comparando-a com dados de outras sequências proteicas, utilizaremos a função Protein BLAST. Também é possível realizar análises de nucleotídeos para nucleotídeos (Nucleotide BLAST), nucleotídeo para proteína (blastx), e proteína para nucleotídeo (tblastn).
Passo 1: Acesse o link da suíte do BLAST – https://blast.ncbi.nlm.nih.gov/Blast.cgi
Passo 2: Selecione a ferramenta Protein BLAST

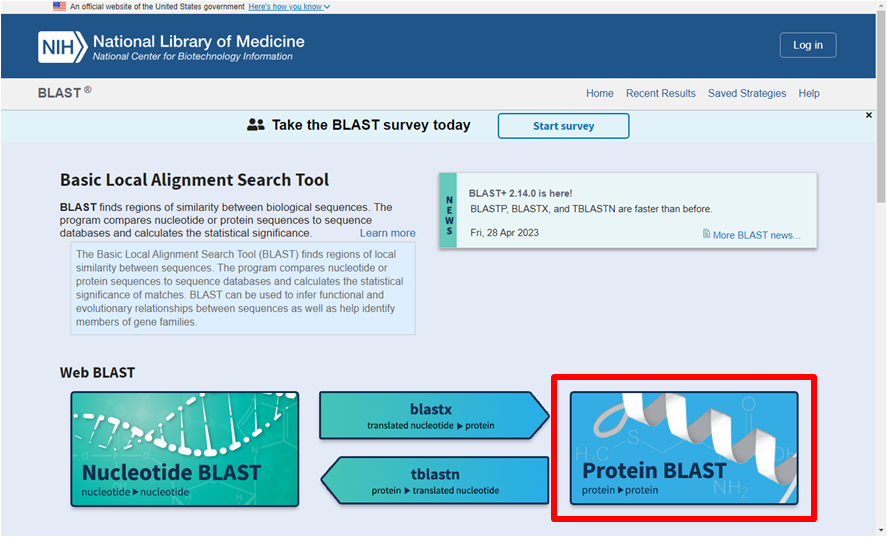
Passo 3: Cole a sequência na caixa localizada na parte superior da página (“Enter Query Sequence”). Também é possível escolher um arquivo com a sequência ou um número de acesso de outro banco de dados (como o Uniprot, por exemplo). Múltiplas sequências simultâneas também são permitidas, desde que estejam em formato FASTA (https://www.ncbi.nlm.nih.gov/genbank/fastaformat/).

Passo 4: Na opção “Database”, dentro de “Choose Search Set”, escolhemos a opção de banco de dados de sequências de proteínas não-redundantes (nr), que deverá ser usada como padrão. Caso você queira buscar apenas por sequências que já tiveram suas estruturas resolvidas, poderá escolher a opção do banco de dados do Protein Data Bank (PDB), por exemplo.

Passo 5: Clique no botão “BLAST”, no fim da página, e aguarde a execução.
Após a execução do programa, a tela de resultados conterá as informações da sua busca na parte superior, e uma lista na parte inferior. Vamos analisar esta lista com mais calma. Caso tenha alguma dúvida, você também pode clicar nas páginas de ajuda, que contam com um tutorial de leitura da página, assim como vídeos no YouTube (a playlist está disponível em https://www.youtube.com/playlist?list=PL7dF9e2qSW0azL2xOKAtxDW7QI8UU4XZ6).

Passo 6: Na aba de descrições, você pode conferir um sumário de todos os “hits” encontrados pelo BLAST. A primeira coluna representa a descrição da proteína encontrada, a segunda o nome da espécie associada, e as outras representam métricas de qualidade do programa.

É possível perceber que todos os “hits” estão ordenados pelo “E-value”, que é a principal métrica de confiança utilizada, e mede a probabilidade de um hit aleatório ocorrer com essa sequência. Ou seja, quanto menor o valor, maior a confiança de que a sua sequência inserida é realmente a informada.
As colunas de cobertura e porcentagem de identidade (à esquerda e direita da coluna de “E-value”, respectivamente), também trazem informações importantes. Suas descrições detalhadas podem ser visualizadas no link: https://blast.ncbi.nlm.nih.gov/doc/blast-help/FAQ.html.
Passo 7: Na aba de alinhamentos, é possível comparar, resíduo a resíduo, a sequência inserida como entrada e aquelas encontradas pela busca do BLAST. Por padrão, essas sequências também estão ordenadas de acordo com o “E-value”.
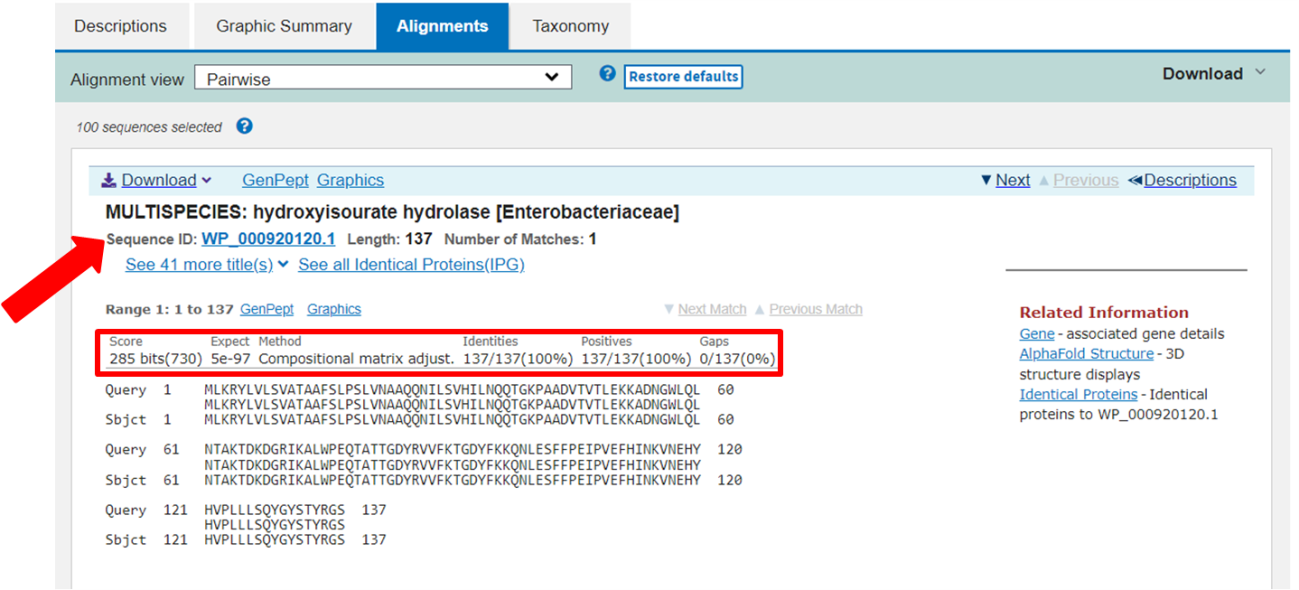
No exemplo acima, podemos perceber que a sequência com a maior confiança possui um “E-value” de 5e-97 (ou seja, bem baixo), 100% de identidade, 100% de alinhamento positivo, e 0% de gaps em relação à nossa sequência de entrada.
Passo 8: De posse da análise do BLAST, e selecionando o melhor resultado obtido, podemos assumir então que nossa sequência de entrada se trata da proteína Hidroxiisourato Hidrolase do gênero de Bactérias Enterobacteriaceae, ou mais especificamente, de Escherichia coli (como pode ser observado nos demais resultados da lista). O identificador da sequência (identificado pela seta vermelha), também é importante, como veremos a seguir.
Passo 9: Copie a sequência identificada na imagem acima (WP_000920120.1).
Mas e agora, que proteína é essa? O que ela faz?
Parte 2 – Uniprot
O Uniprot é um banco de dados mantido pelo Instituto Europeu de Bioinformática (EBI), e que contém informações sobre sequências, estruturas, funções e anotações biológicas de proteínas.
Você pode pesquisar por diversas palavras chave, como o próprio nome ou sigla da proteína, algum organismo de preferência, ou por um número de acesso individual. O número de acesso principal do Uniprot é padronizado no formato de um prefixo “P”, seguido de seis caracteres (ex: P123456), mas diversos outros formatos também são aceitos (como o que acabamos de copiar do BLAST, no passo 9 da parte anterior).
Passo 1: Acesse o link do Uniprot (https://www.uniprot.org/). A tela inicial é mostrada abaixo.

Passo 2: Como acabamos de fazer uma análise no BLAST, nossa primeira tarefa agora é identificar e entender mais sobre os resultados obtidos. Para isso, vamos copiar o código do identificador da sequência com maior confiança (WP_000920120.1), e colá-lo e pesquisá-lo no Uniprot.

Como você pode ter percebido, 6 resultados foram encontrados, mas o primeiro é diferente dos demais por algumas razões:
-
O primeiro resultado possui um código de acesso padrão do Uniprot (P76341), ou seja, esta proteína foi revisada e anotada no SwissProt. Apenas sequências de alta qualidade e confiáveis possuem essa característica.
-
O score de anotação em relação aos demais é superior (5/5), o que demonstra alta evidência experimental dessa proteína. Mais informações sobre os scores de anotação do Uniprot podem ser encontrados aqui: https://www.ebi.ac.uk/training/online/courses/uniprot-exploring-protein-sequence-and-functional-info/annotation-score/
-
A quantidade de informações sobre estruturas 3D e publicações a respeito dessa proteína é superior às demais, evidenciando a maior quantidade de dados biológicos disponíveis.
Passo 3: Clique no primeiro resultado, e você será redirecionado para a página completa dessa proteína, contendo todas as informações disponíveis no UniProt sobre a mesma.

Aqui você pode encontrar informações sobre a função, mecanismo enzimático (apenas para enzimas), sítios de ligação e de modificações pós-traducionais (fosforilações, glicosilações, etc.), taxonomia, localização celular, domínios e outras anotações biológicas. Voltaremos aqui algumas vezes ao longo deste artigo, então não feche a aba. Enquanto isso, fique à vontade para explorar!
Agora que sabemos que nossa sequência desconhecida pertence a uma enzima da via de degradação do ácido úrico em E. coli, quais outras informações podemos obter?
Parte 3 – Características Físico-Químicas
Além do conhecimento da função e outras propriedades que obtivemos a partir do Uniprot, informações físico-químicas das proteínas também são extremamente importantes para uma variedade de aplicações na Bioinformática.
Para esta etapa, utilizaremos a plataforma ExPASy (Expert Protein Analysis System; disponível em: https://www.expasy.org/)[3], mantida pelo Instituto Suíço de Bioinformática (SIB). Essa plataforma disponibiliza gratuitamente diversas ferramentas para ajudar na análise e caracterização de proteínas.

Como não conseguiremos cobrir todas as ferramentas disponíveis (e nem todas são aplicáveis a todos os casos e perguntas), focaremos apenas em uma, embora todas tendam a manter o mesmo padrão de uso. Fique à vontade para explorar as outras!
O ProtParam [4] é uma das ferramentas mais simples disponíveis no ExPASy, mas que fornece informações físico-químicas cruciais para o estudo de proteínas específicas. A documentação da ferramenta pode ser acessada aqui: https://web.expasy.org/protparam/protparam-doc.html.
Passo 1: Acesse o link do ProtParam (https://web.expasy.org/protparam/).

Passo 2: Insira o código Uniprot obtido no passo anterior (P76341) OU a sequência da proteína nas caixas iniciais, e clique em “compute parameters”.
Nota: caso você insira o código Uniprot, uma tela aparecerá para que você selecione qual porção da sequência da proteína quer analisar. É possível perceber que a proteína madura compreende apenas os resíduos de 24 a 137, com os 23 primeiros correspondendo a um peptídeo sinal.

Por curiosidade, podemos ir no Uniprot conferir a informação da localização do peptídeo sinal. Na aba lateral “PTM/Processing”, o primeiro resultado indica justamente a divisão da sequência da proteína em uma parte sinal (resíduos 1-23) e a cadeia da proteína em si (resíduos 24-137). Obviamente, nem todas as proteínas já terão todas suas informações anotadas no Uniprot, por isso a importância de também se realizarem análises teóricas como as que estamos vendo aqui.

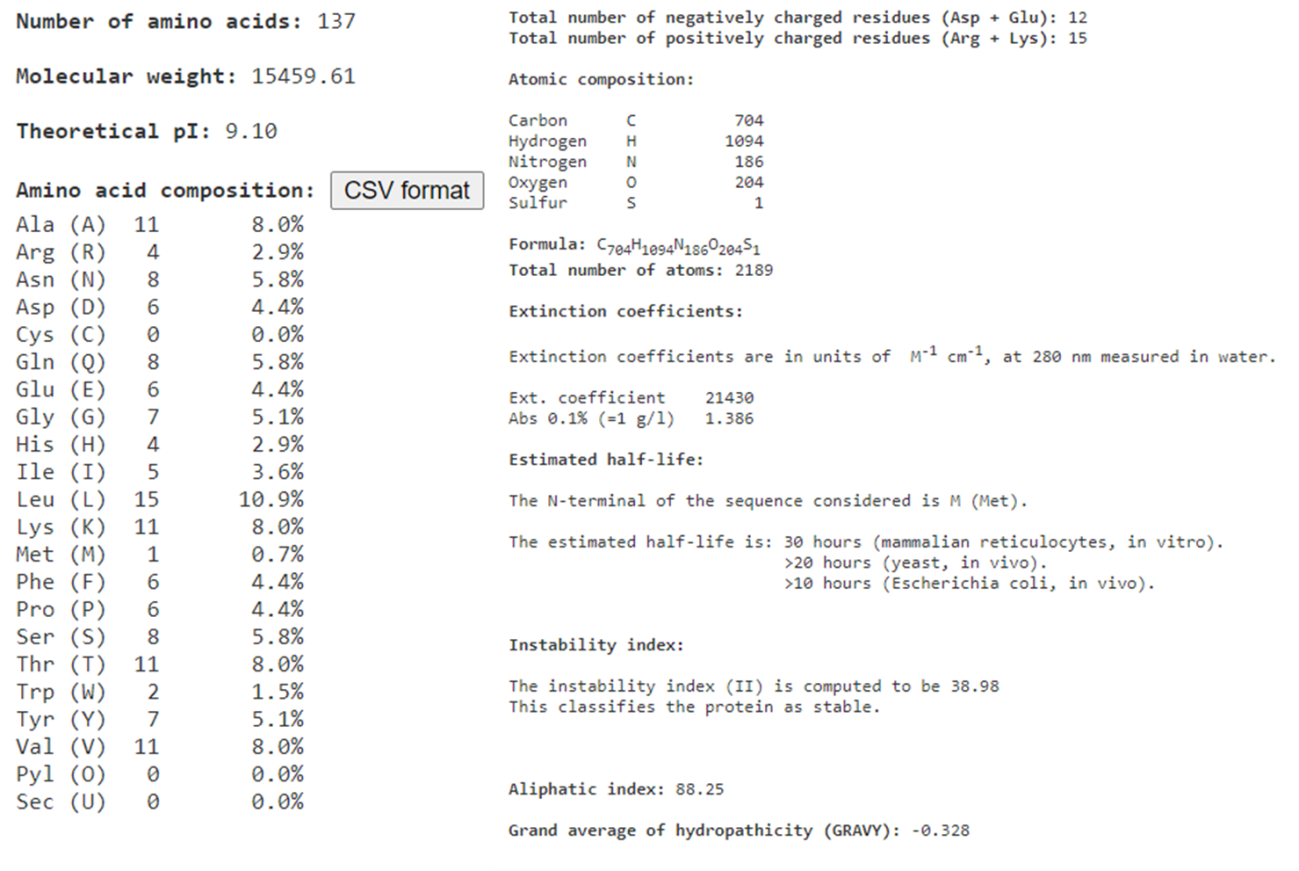
Por agora, apenas clique em “SUBMIT” no final da página do ProtParam, para que a análise seja feita com a sequência completa de 137 resíduos (a mesma que utilizamos para fazer as análises do BLAST).

A janela de resultados do ProtParam mostra informações teóricas como o peso molecular da proteína, ponto isoelétrico (pI), composição atômica e de aminoácidos. Além disso, são calculados o coeficiente de extinção molar, meia-vida da proteína, e índices de estabilidade e hidropaticidade, parâmetros bastante úteis em ensaios experimentais.
Além de parâmetros físico-químicos, que podem ser obtidos diretamente a partir da sequência, também é possível predizer de forma prática a estrutura secundária de uma proteína apenas a partir dessa informação.
Parte 4 – Predição de Estruturas Secundárias
A análise de estruturas secundárias serve como a base do entendimento estrutural de proteínas, e pode fornecer informações importantes apenas com base em uma sequência. Normalmente, estruturas secundárias são divididas em α-hélices, folhas-β e espirais aleatórias [5].
Existem diversas ferramentas para predição computacional de estruturas secundárias, e utilizaremos aqui o servidor web PSIPRED Workbench [6,7].
Passo 1: Acesse o link do PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/)
O PSIPRED fornece uma gama de análises disponíveis, utilizando apenas uma sequência proteica como entrada. Algumas delas são a predição de estruturas secundárias, a predição de desordem estrutural, análises de contatos, reconhecimento de padrões de dobras e de domínios estruturais. Pode-se obter mais detalhes sobre cada análise passando o cursor em cima de cada opção, ou acessando o tutorial da própria ferramenta (http://bioinfadmin.cs.ucl.ac.uk/UCL-CS_Bioinformatics_Server_Tutorial.html).
Como algumas das análises demandam algum tempo e/ou dependem de perguntas biológicas mais específicas, realizaremos neste tutorial apenas a predição de estruturas secundárias, predição de hélices transmembrana e de desordem estrutural (essas duas últimas análises serão mencionadas em mais detalhes na próxima parte). Fique à vontade para explorar as outras opções!
Passo 2: Selecione a opção do PSIPRED 4.0, para predição de estruturas secundárias, MEMSAT-SVM para predição de hélices transmembrana, e DISOPRED3 para predição de desordens estruturais.

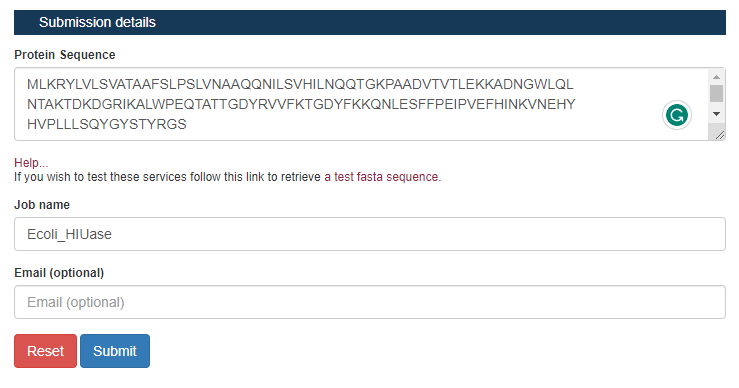
Passo 3: Na aba de “Submission details”, insira a sequência da proteína que estamos trabalhando, um nome para a análise (de preferência algum que torne mais fácil você relembrar o que foi feito), e um email para que os resultados sejam enviados (esta parte é opcional mas recomendada caso você faça análises mais demoradas).
Passo 4: Após finalizadas as análises, os resultados serão mostrados diretamente na ferramenta. Na primeira caixa (“sequence plot”), você pode selecionar para que os dados de uma determinada análise sejam mostrados. No exemplo abaixo, é mostrada a visualização dos resultados do próprio PSIPRED na sequência.
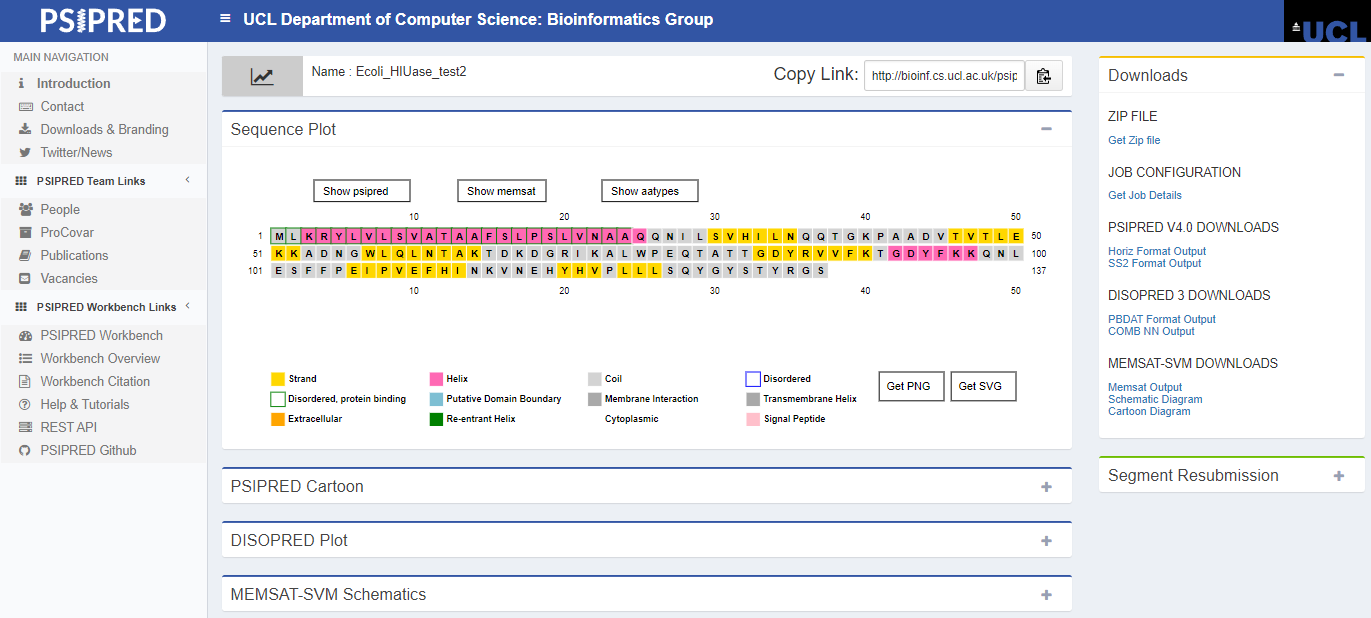
Passo 5: Expandindo cada uma das caixas inferiores, você pode visualizar os resultados individuais de cada análise. Por ora, iremos analisar os resultados da predição de estruturas secundárias em nossa proteína.

Como pode ser percebido, aparentemente temos duas α-hélices (caixas rosas) e sete folhas-β (caixas amarelas). O restante da estrutura é composto por espirais aleatórias (linha cinza). As barras azuis em cima das caixas indicam a confiança de predição aminoácido-específica (quanto mais alta e mais escura a barra, mais alta a confiança).
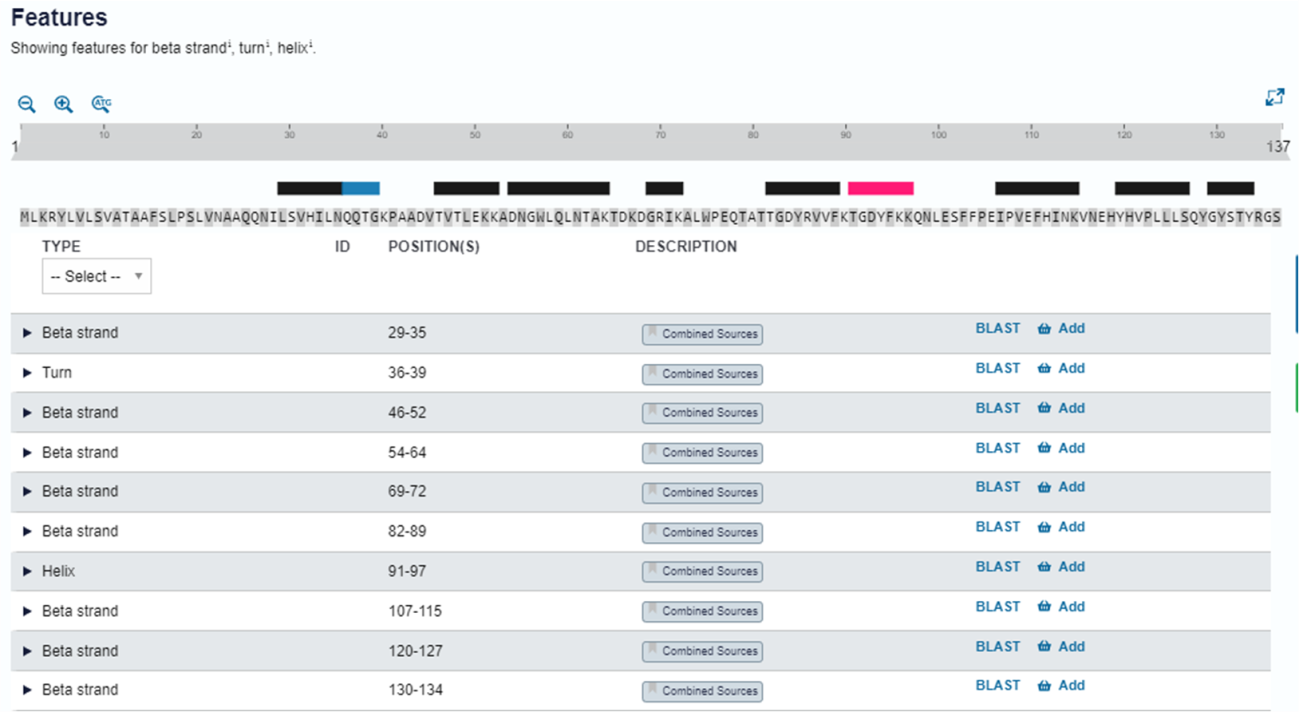
Passo 6: Já que nossa proteína é bem anotada no Uniprot, podemos comparar os dados obtidos de forma teórica pelo PSIPRED com aqueles já depositados e revisados. Na aba lateral “Structure” do Uniprot, na seção “Features”, podemos analisar as estruturas secundárias obtidas experimentalmente para essa proteína, além de identificar a origem dessas informações (clicando nos botões “combined sources” em cada entrada).
Parte 5 – Identificação de Desordem Estrutural e Domínios Transmembrana
Além de informações como a estrutura secundária, também é possível predizer computacionalmente potenciais regiões desordenadas ou transmembranares apenas a partir de uma sequência proteica.
Nesta parte, usaremos os dois outros resultados obtidos anteriormente pelo PSIPRED em conjunto com a ferramenta DeepTMHMM [8], que utiliza deep learning para predizer regiões transmembranares.
Passo 1: No PSIPRED, abra a caixa do resultado do DISOPRED.
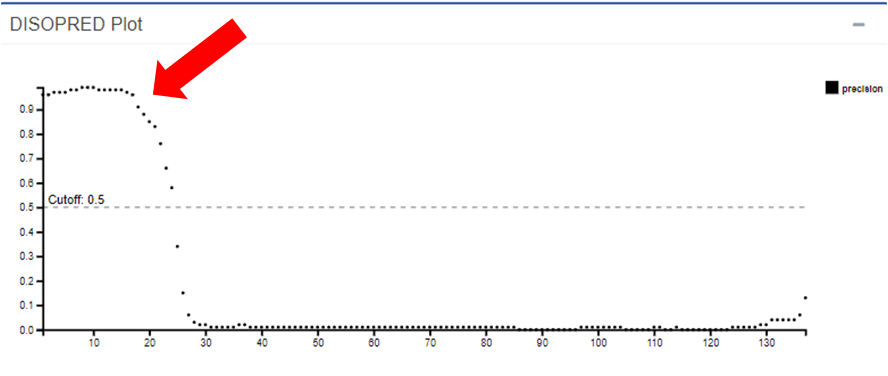
É possível identificar uma região com alta taxa de desordem na proteína (seta vermelha). Essa região pertence justamente ao peptídeo sinal presente nos primeiros resíduos da HIUase. Como peptídeos sinal normalmente são clivados após o endereçamento de uma proteína, essas regiões tendem a ser intrinsecamente desordenadas.
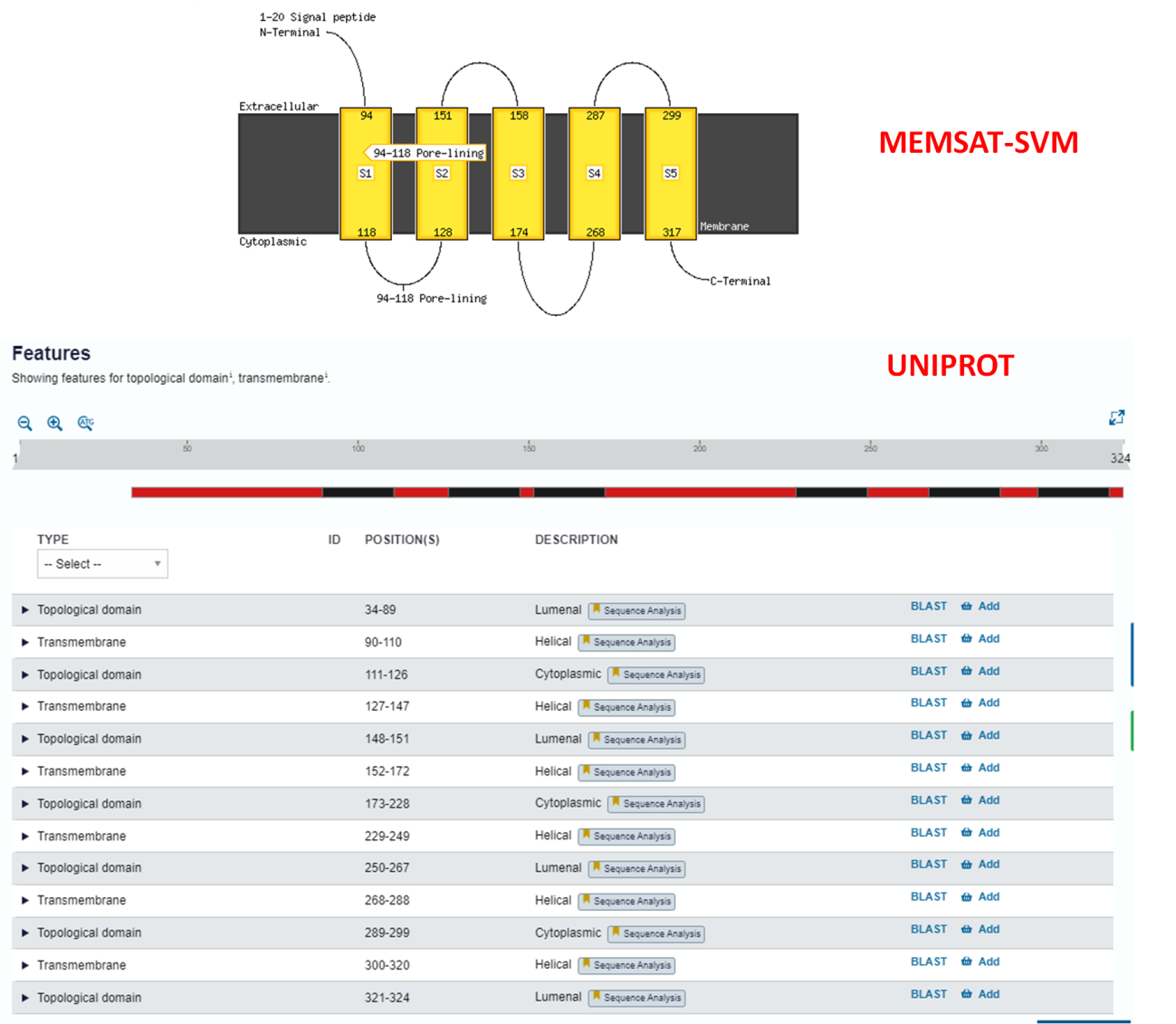
Passo 2: Abra a caixa de resultado do MEMSAT-SVM.

Para esta análise, é possível perceber que a ferramenta identificou os primeiros resíduos da sequência como extracelulares (caixa amarela), além de uma possível região transmembranar/poro entre os resíduos 31 e 46 (caixa preta e azul, e esquema à direita). O peptídeo sinal não foi identificado.
Se olharmos no Uniprot como anteriormente, não há nenhuma menção à regiões transmembranares nessa proteína, assim como regiões extracelulares (a proteína se localiza no periplasma celular, como é possível ver na seção “Subcellular Location” do Uniprot). Sendo assim, a predição desta proteína pela ferramenta MEMSAT-SVM não foi acurada.
Nota: analisando uma proteína verdadeiramente transmembranar (Proteína Transmembranar 165, de Homo sapiens – ID Uniprot: Q9HC07), os resultados são acurados. Para acessar a sequência dessa proteína, use o seguinte link: https://rest.uniprot.org/uniprotkb/Q9HC07.fasta
Como podemos então obter predições mais acuradas sobre regiões transmembranares, tendo em mãos apenas a sequência da proteína?
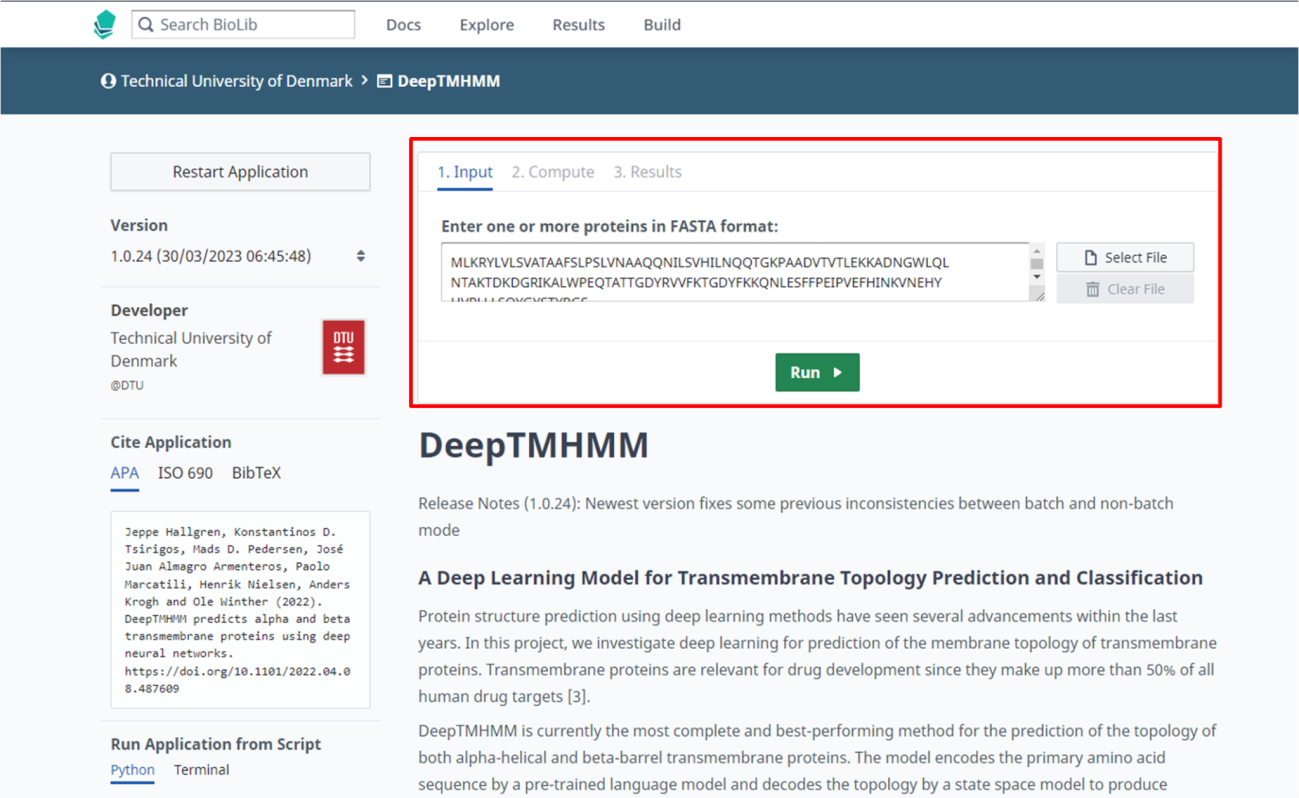
Passo 3: Abra o link do DeepTMHMM (https://dtu.biolib.com/DeepTMHMM).
Passo 4: Insira a sequência da proteína a ser analisada, e clique em “Run”.
Passo 5: Desta vez, os resultados se mostram muito mais condizentes com os dados encontrados no Uniprot, evidenciando a necessidade da utilização de múltiplas ferramentas complementares para obtenção de dados computacionais, especialmente teóricos.
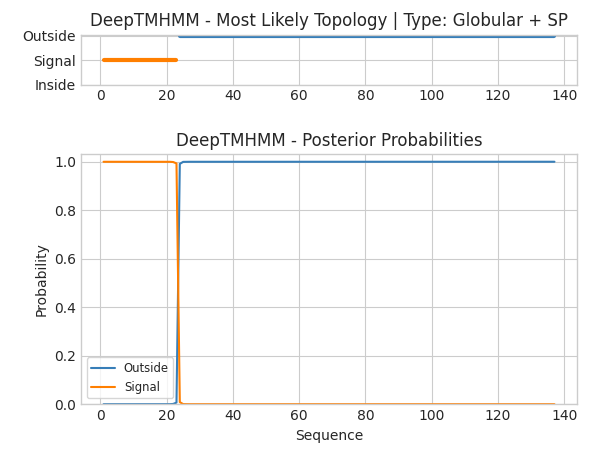
A linha laranja indica região de peptídeo sinal, e a azul indica uma região externa à uma membrana (ou seja, citoplasmática). A topologia da proteína também é predita como globular acompanhada de um peptídeo sinal (SP).
Passo 6: Podemos também realizar uma análise no DeepTMHMM com uma proteína verdadeiramente transmembranar (Q9HC07, a mesma utilizada anteriormente).

Desta vez, além das linhas laranja e azul (peptídeo sinal e região externa, respectivamente), também podemos identificar regiões internas à membrana (linha rosa), e transmembranares (manchas vermelhas). A topologia identificada para a proteína nesse caso é de α-hélices transmembranares, além de um peptídeo sinal.
Parte 6 – Obtenção de Estruturas Tridimensionais
A obtenção de estruturas tridimensionais de proteínas pode ser feita através de diferentes metodologias experimentais ou computacionais [9,10]. Ao resolver as estruturas, os pesquisadores depositam as coordenadas espaciais dos átomos em um banco de dados chamado PDB (Protein Data Bank). Lá você pode ter acesso a uma variedade de estruturas de proteínas depositadas pela comunidade científica ao seu dispor, para utilizá-las em suas análises.
Neste banco de dados é possível fazer a busca utilizando o código da estrutura, que possui como padrão 4 dígitos alfanuméricos (ex: 2H1X, 3IWU, 7KCN, etc). Há também como filtrar pela função da proteína, como Phosphatase, HIUase, Glycosidase, etc. Após realizar a busca você vai receber diversas estruturas com várias informações.
Passo 1: No Uniprot que estamos trabalhando, na seção “Structure”, você pode encontrar todas as estruturas tridimensionais que já foram resolvidas para essa determinada proteína. Não se preocupe se nenhuma estrutura já tiver sido depositada no PDB. Falaremos do AlphaFold mais à frente!
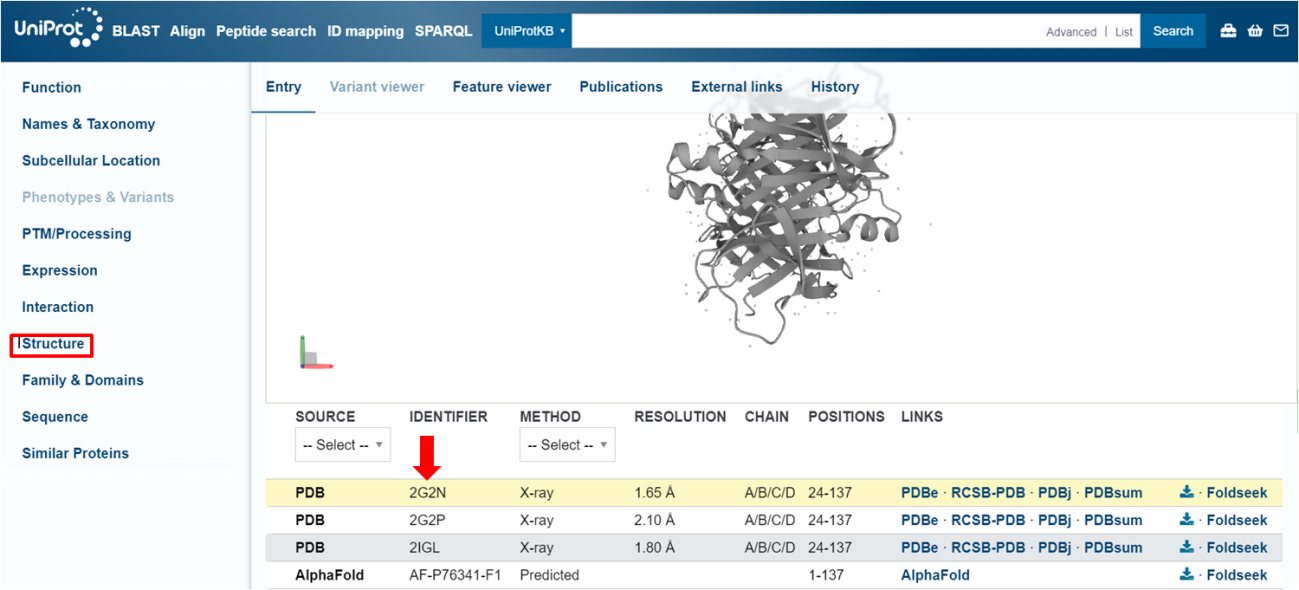
Procure pelo código do PDB da estrutura com melhor resolução. Voltaremos a trabalhar com esse código daqui a pouco.
Passo 2: Acesse o banco de dados do PDB (https://www.rcsb.org/).
Passo 3: Antes de pesquisarmos nossa proteína, vale conferir a parte de busca avançada que pode ser vista abaixo da região de busca.
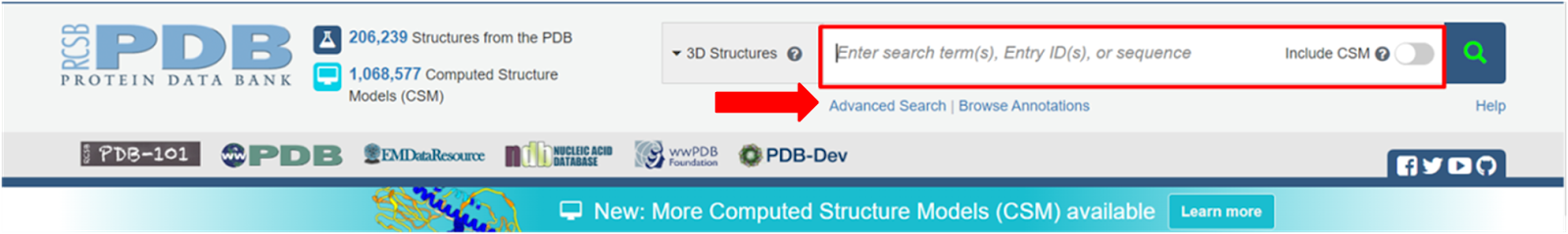
Passo 4: Na busca avançada existe a possibilidade de especificar detalhes estruturais, experimentais e químicos desejados a fim de filtrar as estruturas desejadas. Outros filtros podem ser acessados também após realizar a busca.

Passo 5: Caso seja feita uma busca pela função, por exemplo, é interessante observar os filtros de métodos experimentais, organismo de origem, taxonomia e a resolução das estruturas. Este último é um parâmetro importante para a validação das estruturas. Então, façam o experimento de buscar por “HIUase” utilizando os filtros de “Refinements” para buscar estruturas de diferentes níveis de resolução, organismos, taxonomia, etc.

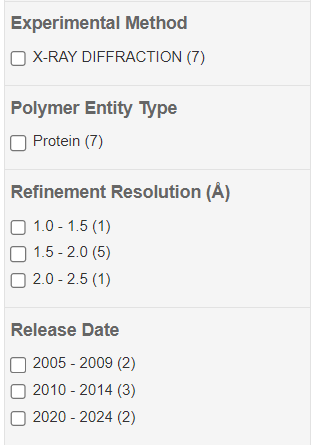
Vale também ressaltar informações importantes para se avaliar nas estruturas, como a resolução em angstroms (quanto menor, melhor), assim como o organismo possuidor da proteína e os ligantes já descritos na literatura.

Passo 6: Voltando para o código PDB que obtivemos anteriormente no Uniprot (2G2N), iremos pesquisá-lo na busca normal do PDB.
Passo 7: Após abrir a estrutura 2G2N, já é possível visualizar diversas informações e validações importantes sobre a estrutura obtida experimentalmente, como os outliers do gráfico de Ramachandran (que será detalhado mais para frente).


Os parâmetros de validação são de extrema importância para a escolha de seu PDB em análises, sejam de dinâmica molecular, modelagem de estruturas ou como objeto de estudo em geral. Ou seja, é um campo que deve ser analisado com cuidado e sendo sempre comparado com outras estruturas similares.
Passo 8: Aqui, também é possível observar parâmetros como hidropaticidade dos resíduos, grau de desordem (mobilidade dos resíduos) e também informações da sequência no Uniprot. Lembre-se que já calculamos a maioria desses parâmetros anteriormente, mas de forma teórica. Fique à vontade para compará-los com as informações experimentais!

Para a obtenção de informações estruturais confiáveis, algumas metodologias experimentais necessitam do uso de pequenas moléculas, seja para estabilizar a estrutura ou para melhorar a resolução do experimento. Algumas perguntas também exigem a obtenção da proteína com seus ligantes biológicos, assim como inibidores. Nesta seção do PDB é possível verificar a presença das moléculas que foram obtidas juntamente à proteína depositada.

Todas as informações sobre a estrutura tridimensional de uma proteína estão disponíveis no formato “.pdb”. Esse é um arquivo de texto (ou seja, pode ser aberto em qualquer editor, como o Word, Notepad, etc.), contendo as coordenadas tridimensionais de cada átomo presente na estrutura. Além disso, há um cabeçalho com informações adicionais, como as condições experimentais para obtenção da estrutura, referências, etc.
Passo 1: Você pode visualizar o arquivo .pdb da proteína clicando em “Display Files” e depois em “PDB Format”. Também é possível baixar o arquivo clicando em “Download Files” e depois em “PDB Format”.
Nota: Para visualizar diretamente o .pdb da estrutura 2G2N, acesse em “https://files.rcsb.org/view/2G2N.pdb”
.

Passo 2: Abra o arquivo .pdb da estrutura 2G2N em algum editor de texto (ou no próprio navegador). No cabeçalho é possível ver algumas informações como o organismo na qual a proteína foi expressa, o sistema de expressão utilizado, assim como a metodologia usada para resolver a estrutura.

Também há informações sobre o artigo publicado referente ao depósito da estrutura no PDB. É interessante lê-lo para obter informações adicionais sobre a proteína.

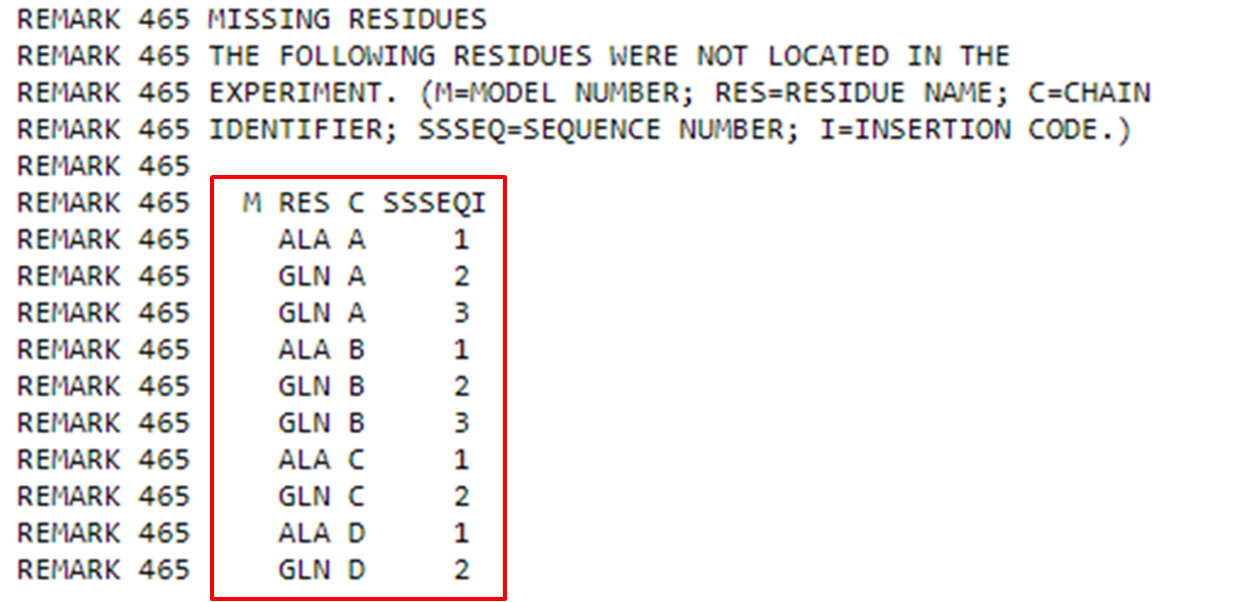
Logo após, na seção de “Remarks”, você pode obter todas as outras informações sobre a proteína (antes das coordenadas tridimensionais). Mais informações sobre os remarks podem ser obtidas aqui: https://www.wwpdb.org/documentation/file-format-content/format33/remarks1.html.
Um remark bastante importante são os “Missing Residues”. É bastante comum que alguns resíduos com grande mobilidade não sejam detectados em metodologias como Cristalografia de Raios X. Mas, note que neste caso se trata apenas dos primeiros resíduos da proteína que possuem graus de liberdade maiores e são mais desordenados. Em seguida também poderemos observar outros parâmetros estruturais.
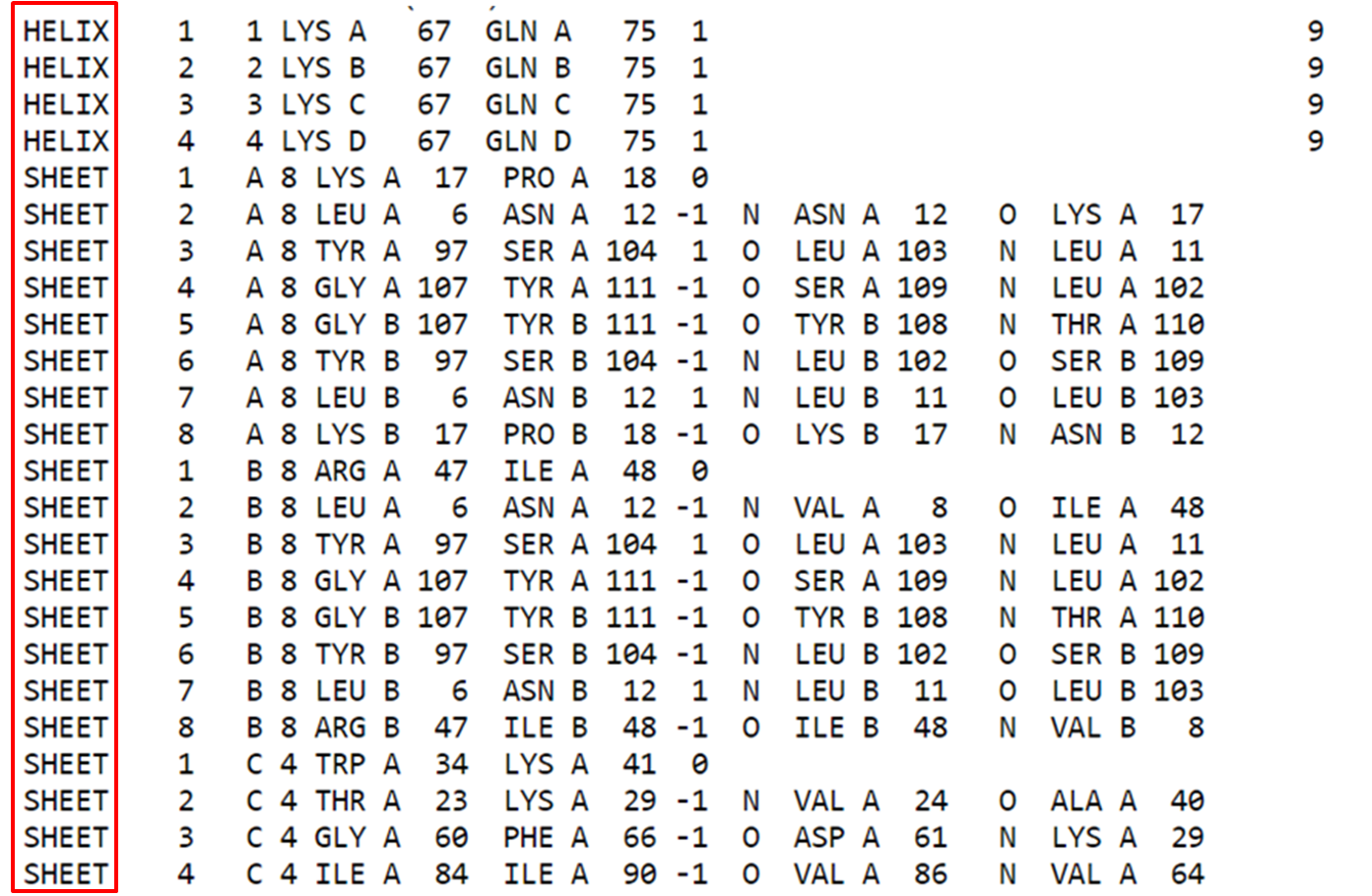
Na parte estrutural (após os remarks), podemos observar informações sobre resíduos que contribuem para estruturas secundárias da proteína, ou seja, aqueles resíduos que formam alfa hélices e folhas beta.
O cabeçalho também pode fornecer informações sobre sítios ativos das proteínas, podendo dar certa noção de resíduos importantes na função proteica.
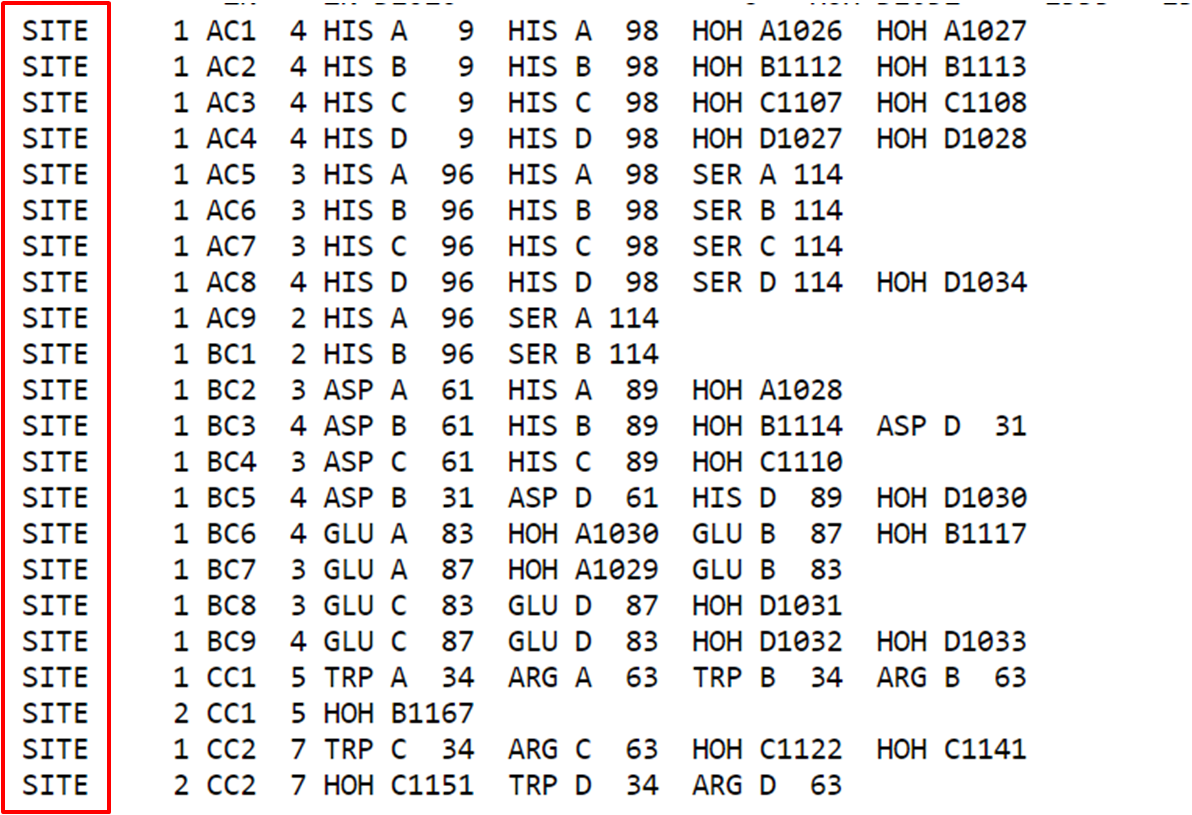
Passo 3: Após o cabeçalho, chegamos na seção de “Atoms”. Em geral as coordenadas dos átomos são dispostas da seguinte forma:
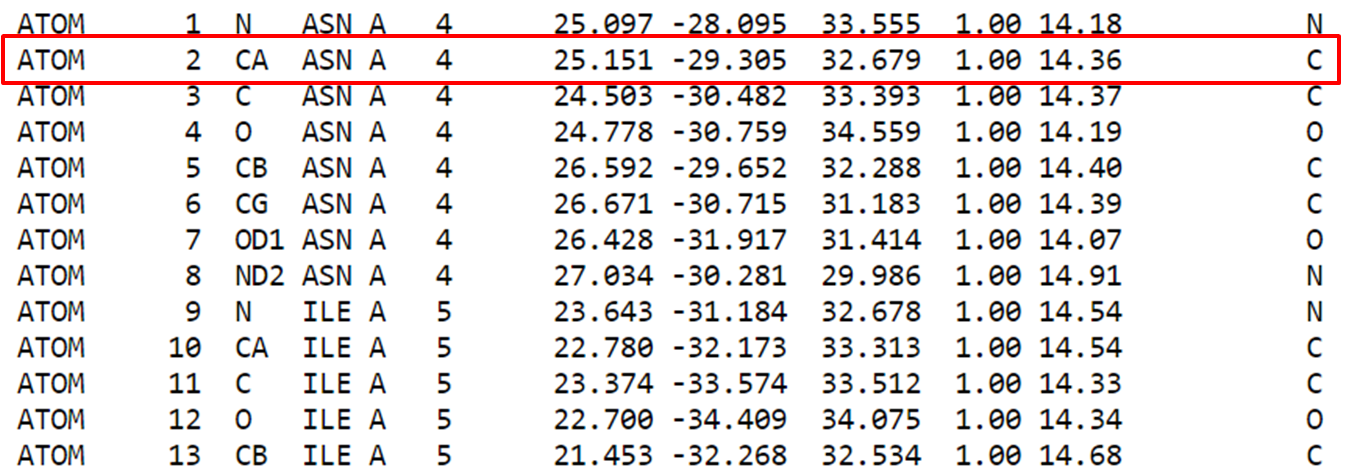
Cada átomo dispõe de diversas informações como: número, tipo atômico, o resíduo e cadeia na qual pertence, a posição do resíduo, as coordenadas cartesianas (x, y e z) e por fim a ocupância e fator-B.
Para exemplificar, tomemos o átomo de carbono marcado em vermelho na figura anterior. Ele é um carbono alfa descrito pelo tipo atômico CA de numeração 2 do resíduo de Asparagina 4 na cadeia A da estrutura. Logo temos as coordenadas x, y e z e em seguida um valor de ocupação 1.00 e B-fator de 14.36. Vale verificar a documentação do formato de arquivo que pode ser obtida em https://www.wwpdb.org/documentation/file-format.
Parte 8 – Validação Estrutural
Após obter a sua estrutura de interesse (seja experimentalmente ou computacionalmente), é importante realizar a validação da mesma. Existem diversos parâmetros que podem ser facilmente analisados através de métodos como os gráficos de ERRAT e de Ramachandran, que serão detalhados nesta parte.
Passo 1: Acesse o servidor SAVES em “https://saves.mbi.ucla.edu/”.

Passo 2: Submeta sua estrutura no formato “.pdb” e selecione “Run programs”.
Passo 3: Iremos utilizar duas das ferramentas do SAVES: a) ERRAT [11], que avalia a estabilidade e confiança estatística da conformação dos resíduos baseando-se em estruturas de referência; b) PROCHECK [12], que nos permite fazer o gráfico de Ramachandran [13], importante ferramenta de validação de estruturas por meio da análise de permissões dos rotâmetros phi (φ) e psi (Ψ) dos resíduos de aminoácidos.

Passo 4: O ERRAT irá retornar um valor de confiança de erro para a estrutura. Estruturas resolvidas experimentalmente tendem a ter valores superiores à 90, como é o caso da estrutura utilizada. Assim como tudo na biologia, exceções existem e devem ser analisadas cuidadosamente.

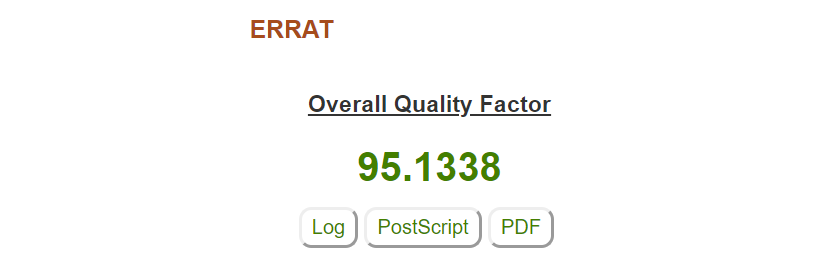
No gráfico do ERRAT é possível observar o nível de confiança de cada resíduo individualmente. Resíduos indicados em amarelo se encontram acima do intervalo de 95% de confiança de rejeição, e resíduos em vermelho acima de 99%. Os resíduos coloridos tendem a se encontrar em regiões de volta na proteína, ou são aqueles que possuem posições mais móveis e instáveis.
Nota: Caso sua proteína tenha mais de uma cadeia (como é o caso do nosso exemplo), o ERRAT irá calcular o gráfico para cada uma individualmente. Mesmo no caso de homotetrâmeros como a HIUase, é possível identificar pequenas variações entre as cadeias.

Passo 5: O PROCHECK pode demorar alguns minutos para ser calculado, mas depois de terminado, clique em “Results”.


O programa pode mostrar avisos e erros que podem ser verificados na aba lateral. Não se preocupe tanto com eles, já que a principal métrica que analisaremos aqui é o gráfico de Ramachandran.


Como podemos ver, o aviso significa que outras avaliações podem ser necessárias para refinar a estrutura. Você pode acessar outras ferramentas que desempenham essas funções, como o PDB-REDO (mais detalhes na última parte deste manuscrito). Sabendo disso, podemos analisar o gráfico de Ramachandran.


Aqui podemos ver as regiões permitidas para a rotação dos ângulos phi e psi, onde as regiões ideais (ou mais permitidas) são aquelas em vermelho. As amarelas e beges são permitidas mas menos aceitas, e em branco são as regiões não permitidas. A quantidade de pontos fora da região vermelha será o seu parâmetro de avaliação da qualidade da estrutura.
No caso da nossa proteína, podemos ver que 92,3% dos resíduos se encontram na região vermelha, 7,4% na região amarela, 0,3% na região bege, e 0% na região branca. Porém, como dito anteriormente, o gráfico de Ramachandran é apenas uma evidência sobre a qualidade da estrutura, e outras análises mais aprofundadas podem se valer necessárias (você pode encontrar recursos adicionais na última parte deste manuscrito).


Parte 9 – Visualização de Estruturas
A partir da estrutura tridimensional obtida, o que fazer? Existem diversas informações que podem ser retiradas de suas estruturas através da inspeção visual, e para isto alguns softwares se destacam, como o ChimeraX [14], que será usado para exemplificação.
Passo 1: Faça o download do programa ChimeraX em “https://www.cgl.ucsf.edu/chimerax/download.html”.
Passo 2: Após instalação, podemos abrir uma estrutura do PDB da seguinte maneira:


Com sua proteína aberta, a interface fica da seguinte forma:


Agora, vamos navegar por algumas funções importantes, mas antes, vamos mudar o fundo.


Passo 3: Em Presets, selecione a opção Publication 1 (Silhouettes).
Passo 4: Na seção Molecule Display podemos acessar a sequência da estrutura carregada.

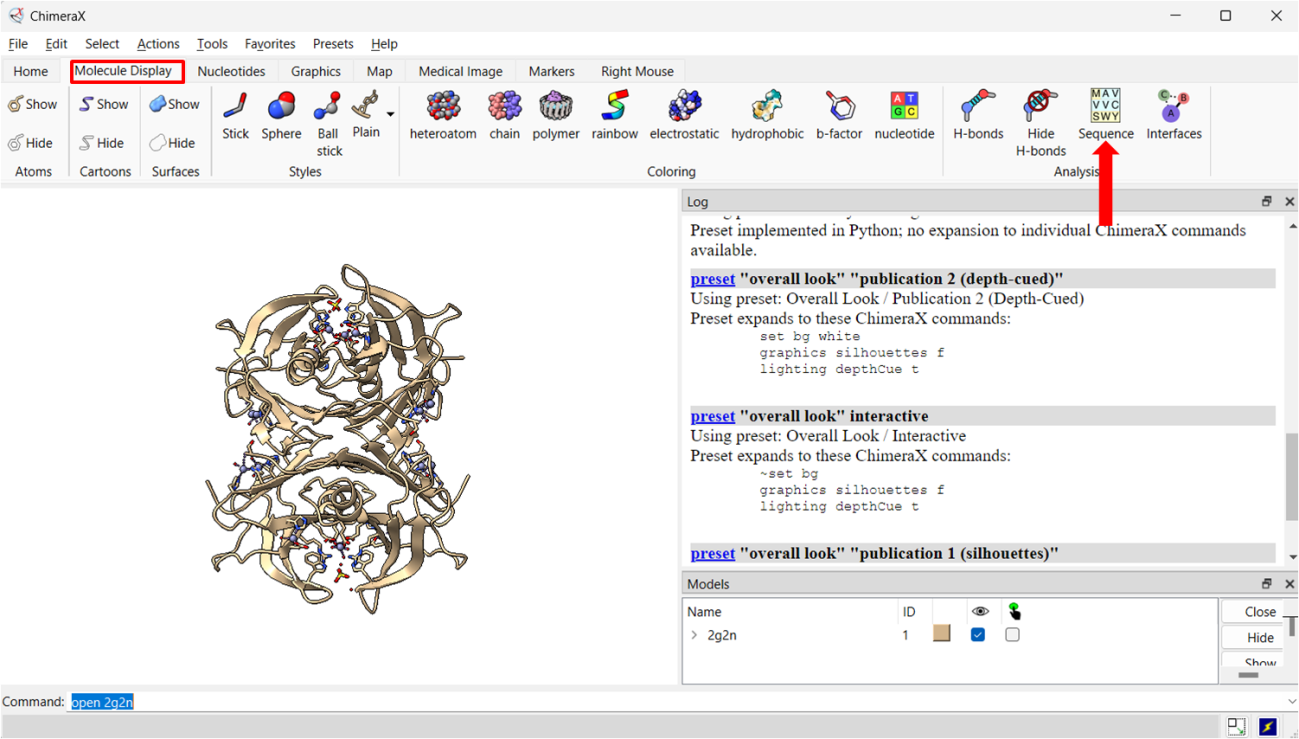


Com o botão esquerdo é possível selecionar resíduos das cadeias, e após selecionado algumas ações podem ser feitas, como mudar a representação gráfica desses resíduos.
Passo 5: Selecione alguns resíduos na sequência e altere a visualização dos mesmos para “Sphere”, e a cor para “Rainbow”.

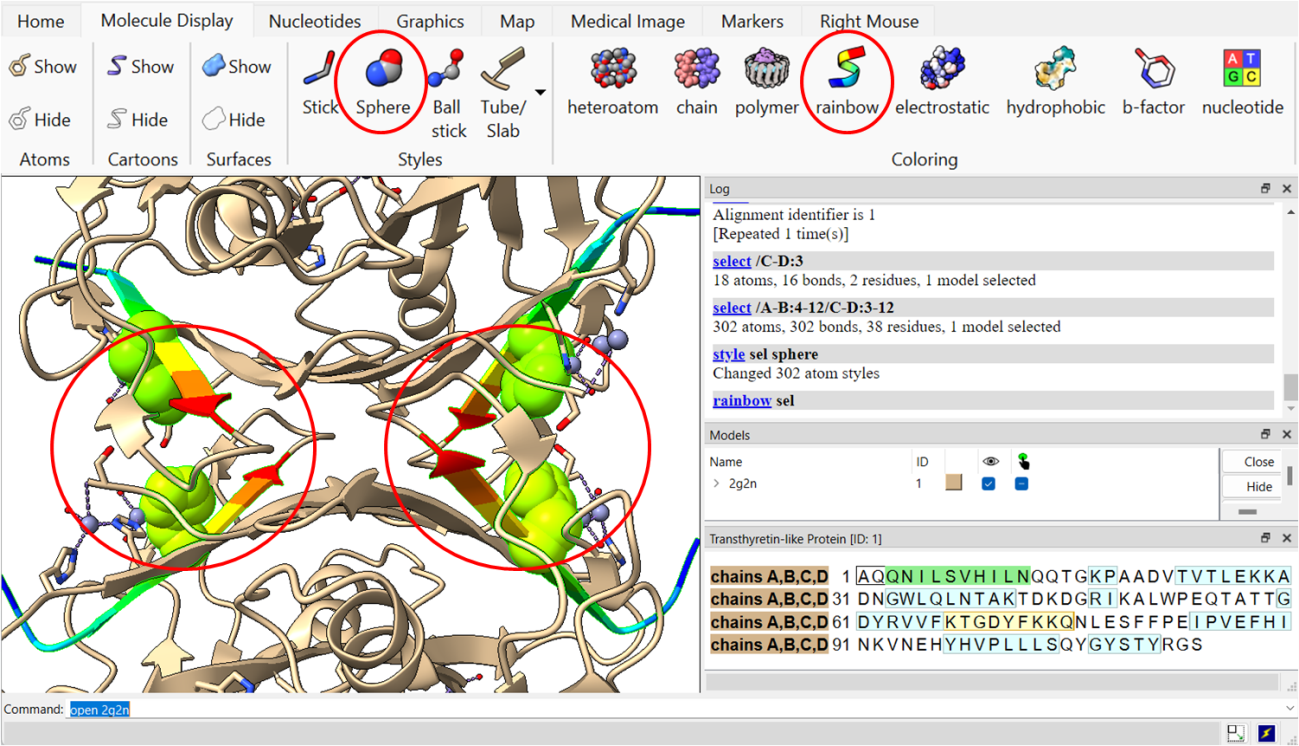
Passo 6: Também é possível colorir toda sua estrutura de acordo com o nível de B-fator dos resíduos, que irá nos dar a informação das regiões de maior mobilidade da estrutura.




Passo 7: Podemos analisar as ligações de hidrogênio (próximo da seção Sequence que foi usada anteriormente) feitas pelos resíduos da proteína.

É possível selecionar aqueles resíduos de interesse como mostrado anteriormente e verificar as interações.

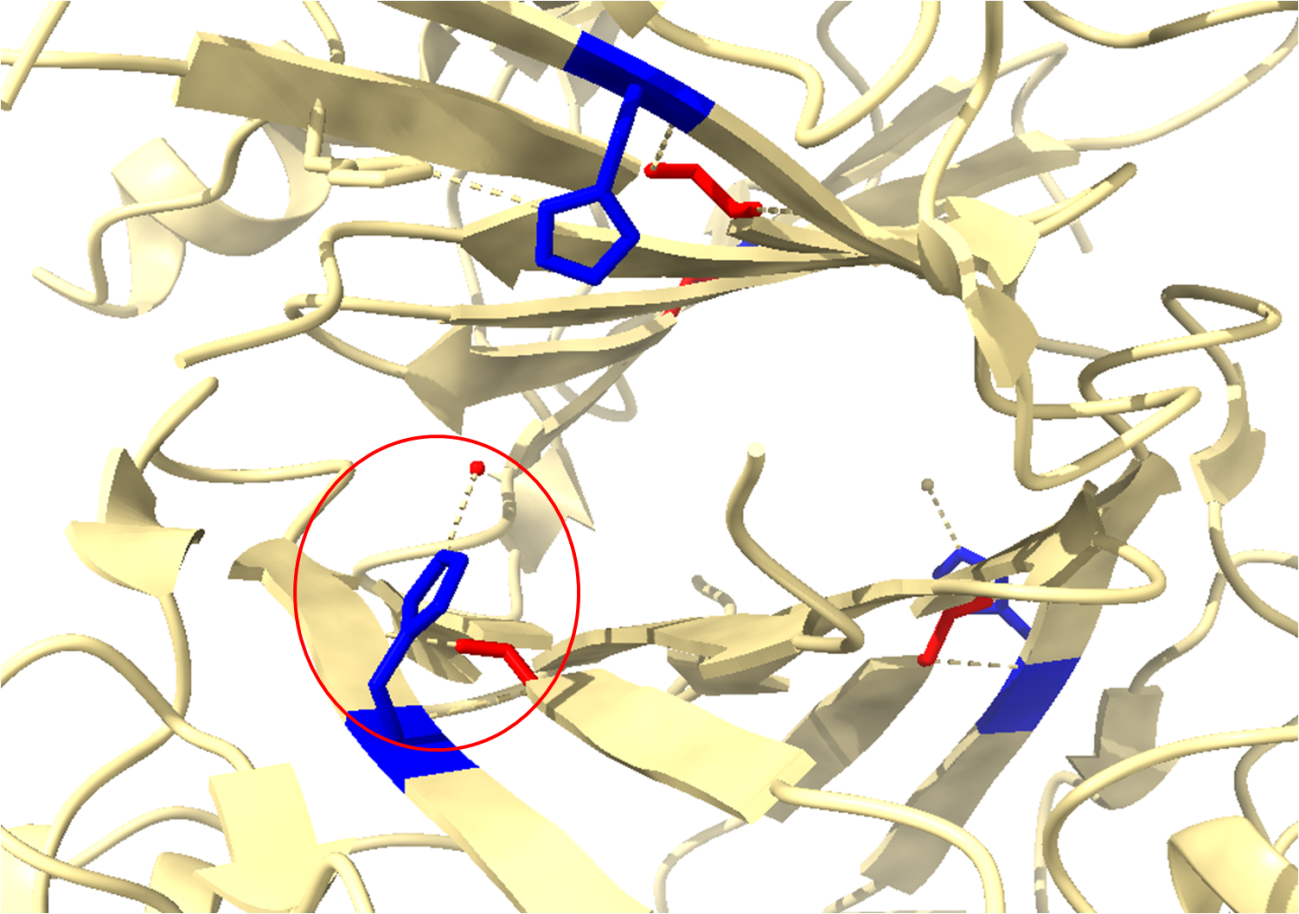
Nesta seleção pode-se observar as interações de hidrogênio dos resíduos 9 (His) e 101(Leu). Note a presença da interação da Histidina com uma molécula de oxigênio representando a água (estruturas cristalográficas não possuem hidrogênios). Esta é uma informação estrutural de extrema importância para esta família de proteínas, visto que a sua ação catalítica parte da transferência de um próton da água para um resíduo de histidina no sítio ativo. E na sua proteína de interesse, quais interações podem ser vistas apenas pela visualização da molécula pelo ChimeraX? Fique à vontade para explorar!
Parte 10 – Introdução ao AlphaFold
Em 2021, foi publicada pela empresa DeepMind a ferramenta AlphaFold, que realiza a predição estrutural e modelagem de proteínas, utilizando apenas a sequência como entrada [15]. Essa ferramenta se baseia em uma abordagem de inteligência artificial, redes neurais, e aprendizado profundo. Mais detalhes podem ser obtidos em um artigo da própria revista BIOINFO [16].
Contudo, nosso foco neste manuscrito não é discutir o AlphaFold em detalhes ou mesmo ensiná-lo a usá-lo para modelar proteínas, e sim discutir as potenciais aplicações da ferramenta e quais informações podem ser aproveitadas de sua predição. Mais informações sobre o AlphaFold podem ser encontradas aqui: https://www.deepmind.com/research/highlighted-research/alphafold.
Nota: Caso você tenha interesse no funcionamento do AlphaFold e quiser testá-lo, foi publicada uma versão do programa aberta, gratuita e disponível em nuvem, denominada ColabFold [17]. O ColabFold utiliza o GoogleColab e possui diversas versões do AlphaFold disponíveis. Você pode acessá-lo por aqui: https://github.com/sokrypton/ColabFold.
Vamos ao que interessa: o AlphaFold se mostrou uma ferramenta tão poderosa para a predição de estruturas de proteínas que não haviam sido ainda depositadas que um grande esforço foi feito para que todas as proteínas do Uniprot fossem resolvidas por meio da ferramenta (isso mesmo, todas as mais de 200 milhões de sequências protéicas conhecidas pela ciência). O resultado se mostrou assustadoramente confiável para a grande maioria delas, mas obviamente nem todos os modelos possuem uma boa qualidade. Mais informações sobre a empreitada podem ser encontradas aqui: https://www.deepmind.com/blog/alphafold-reveals-the-structure-of-the-protein-universe
E agora, como saber se os modelos são confiáveis ou não?
Passo 1: Acesse novamente o ID Uniprot da proteína que estamos trabalhando (P76341). Na aba lateral esquerda, clique em “Structure”.

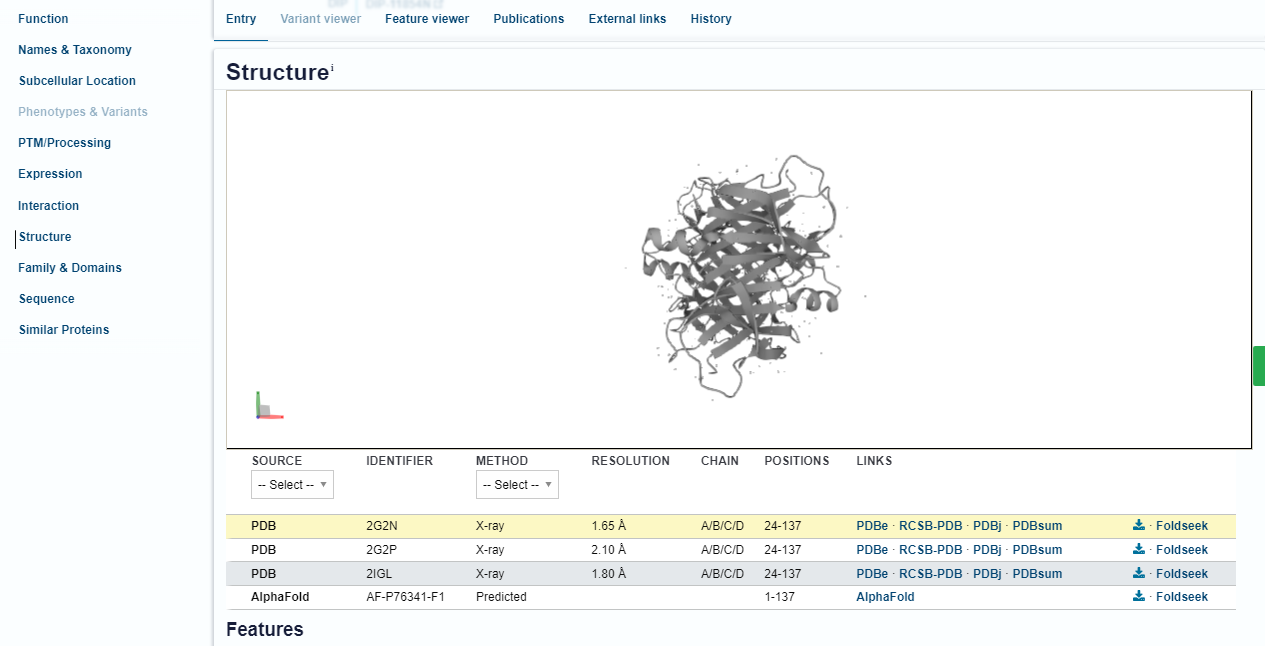
Como nossa estrutura já possui modelos experimentais depositados no PDB, você deve perceber, abaixo da visualização da estrutura, que existem diversas entradas. Inclusive, a primeira delas é a 2G2N, que acabamos de trabalhar e analisar nos passos anteriores. Você pode identificar a origem, ID, método de obtenção, resolução, cadeias e resíduos, etc.
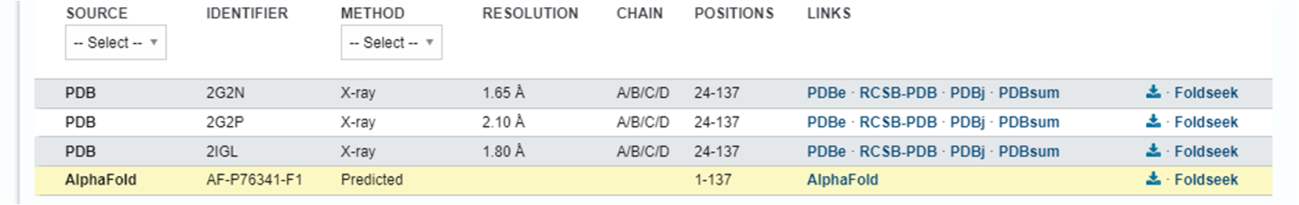
Passo 2: Suponhamos que sua proteína de interesse ainda não foi resolvida experimentalmente. Dessa forma, apenas a entrada modelada computacionalmente pelo AlphaFold estará disponível. Clique nela.


À esquerda da visualização da proteína, temos agora a principal métrica de confiança do AlphaFold, chamada pLDDT (“Predicted Local Distance Difference Test”)[18]. Essa métrica baseia-se em uma avaliação resíduo-específica da distância local entre todos os átomos, e como o ambiente de uma estrutura de referência é reproduzido em um modelo.
Valores azuis escuros de pLDDT (>90) indicam alta confiança na modelagem daquele resíduo específico, enquanto valores laranjas indicam baixíssima confiança (<50). Na nossa proteína, é possível observar que quase toda a estrutura do monômero se encontra azul escura (ou seja, modelada com alta confiança), enquanto uma pequena porção dos resíduos possui coloração amarelada/alaranjada. Esses últimos resíduos se referem justamente aos primeiros resíduos da sequência, que formam o peptídeo sinal e não possuem uma conformação definida. É importante lembrar que apenas uma das 4 cadeias da proteína foi modelada!
Nota: Você pode brincar com a visualização da proteína na própria página do Uniprot utilizando a roda do mouse para dar zoom e clicando e arrastando para rotacionar. Ao passar o mouse em cima de algum resíduo, as informações do mesmo irão aparecer no canto inferior direito.
Passo 3: Para obter mais informações sobre o modelo do AlphaFold, clique no link do AlphaFold disponível no próprio Uniprot.

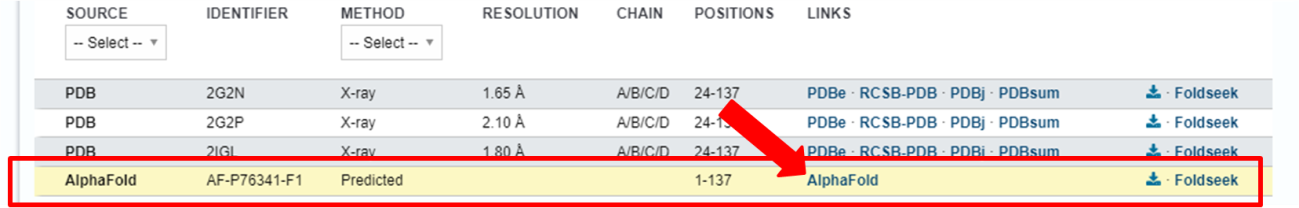
Aqui, além de poder visualizar melhor a estrutura da proteína, você pode obter outras informações de qualidade, como a métrica de PAE (“Predicted Alignment Error”), que mede a posição relativa das cadeias da proteína, utilizando a distância entre os resíduos. O gráfico de PAE é mostrado logo após a estrutura da proteína, e mais informações e interpretações podem ser obtidas logo abaixo, na caixa de tutorial.

Passo 4: Se quiser analisar a estrutura em alguma ferramenta externa (como ChimeraX ou PyMOL), baixe o arquivo PDB da proteína modelada pelo AlphaFold.

Parte 11 – Ferramentas Extras
Por fim, apresentaremos, em ordem alfabética, outras ferramentas online e gratuitas que podem ser úteis para investigação computacional de sequências e estruturas de proteínas.
-
CATH: Base de dados de classificação de estruturas e domínios proteicos do Protein Data Bank em superfamílias. Disponível em: http://cathdb.info/.
-
CAVER: Programa para análise e visualização de cavidades e túneis em estruturas de proteínas. Possui uma versão para download (CAVER Analyst), e uma versão online como plugin para o visualizador de estruturas PyMOL. Disponível em: https://www.caver.cz/.
-
ClustalOmega: Ferramenta para alinhamento múltiplo de sequências. Disponível em: https://www.ebi.ac.uk/Tools/msa/clustalo/.
-
HMMER: Realiza alinhamentos de sequências e busca em bancos de dados por sequências homólogas de proteínas. O AlphaFold utiliza uma versão modificada (chamada jackHMMER) para realizar a busca de sequências para modelagem. Disponível em: http://hmmer.org/.
-
PDB REDO – Esta plataforma valida e oferece versões otimizadas das estruturas depositadas no PDB, alterando certos parâmetros a fim de obter estruturas que melhor representem as formas ativas das proteínas. Disponível em: https://pdb-redo.eu/.
-
PDB Sum – Fornece um “resumo” de uma entrada no PDB. Você pode obter informações rapidamente, como estrutura secundária, sítios de ligação, cavidades, etc. Disponível em: https://www.ebi.ac.uk/thornton-srv/databases/pdbsum/.
-
PFAM/InterPro: Base de dados sobre famílias proteicas. Você pode obter informações como domínios, proteínas relacionadas por similaridade de sequências ou características funcionais, etc. Disponível em: https://www.ebi.ac.uk/interpro/.
-
PreStO – Webservice para encontrar ferramentas computacionais relacionadas à biologia estrutural. Você pode procurar por termos específicos ou na lista de quase 100 ferramentas listadas. Disponível em: http://bioinfo.dcc.ufmg.br/presto/.
-
PROPKA – Ferramenta de predição de estados de protonação. O programa auxilia na escolha de protonação dos resíduos de uma proteína visto que algumas metodologias de resolução de estruturas, como a cristalografia, não resolvem a posição dos hidrogênios. Um pipeline oferecido pela plataforma prediz os valores de pKa dos resíduos e retorna um arquivo PDB com a protonação no pH desejado. Disponível em: https://server.poissonboltzmann.org/pdb2pqr.
-
SCOPe – Base de dados de classificação de estruturas proteicas. Disponível em: https://scop.berkeley.edu/.
-
SMART: Similar ao PFAM, fornece informações como estruturas de domínios e famílias proteicas. Disponível em https://smart.embl-heidelberg.de/.
-
VADAR – Ferramenta especialmente direcionada para a identificação de resíduos expostos ou enterrados em estruturas. Diversas outras informações, como estruturas secundárias e métricas de qualidade também podem ser obtidas. Disponível em: http://vadar.wishartlab.com/.
Referências
1. Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410, doi:10.1016/S0022-2836(05)80360-2.
2. Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402.
3. Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The Proteomics Server for in-Depth Protein Knowledge and Analysis. Nucleic Acids Res. 2003, 31, 3784–3788.
4. Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Springer Protocols Handbooks; Humana Press: Totowa, NJ, 2005; pp. 571–607 ISBN 978-1-59259-890-8.
5. Lyu, Z.; Wang, Z.; Luo, F.; Shuai, J.; Huang, Y. Protein Secondary Structure Prediction With a Reductive Deep Learning Method. Front. Bioeng. Biotechnol. 2021, 9, 687426, doi:10.3389/fbioe.2021.687426.
6. McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED Protein Structure Prediction Server. Bioinformatics 2000, 16, 404–405, doi:10.1093/bioinformatics/16.4.404.
7. Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 Years On. Nucleic Acids Res. 2019, 47, W402–W407, doi:10.1093/nar/gkz297.
8. Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Armenteros, J.J.A.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM Predicts Alpha and Beta Transmembrane Proteins Using Deep Neural Networks 2022, 2022.04.08.487609.
9. Silva, Letícia Xavier; Bastos, Luana Luiza; Santos, Lucianna Helene. Modelagem computacional de proteínas. In: BIOINFO – Revista Brasileira de Bioinformática e Biologia Computacional, v.1, 2021, n.8. doi: 10.51780/978-6-599-275326-08
10. Schaffer, J.E.; Kukshal, V.; Miller, J.J.; Kitainda, V.; Jez, J.M. Beyond X-Rays: An Overview of Emerging Structural Biology Methods. Emerg. Top. Life Sci. 2021, 5, 221–230, doi:10.1042/ETLS20200272.
11. Colovos, C.; Yeates, T.O. Verification of Protein Structures: Patterns of Nonbonded Atomic Interactions. Protein Sci. 1993, 2, 1511–1519, doi:10.1002/pro.5560020916.
12. Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291, doi:10.1107/S0021889892009944.
13. Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of Polypeptide Chain Configurations. J. Mol. Biol. 1963, 7, 95–99, doi:10.1016/S0022-2836(63)80023-6.
14. Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. Publ. Protein Soc. 2021, 30, 70–82, doi:10.1002/pro.3943.
15. Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589, doi:10.1038/s41586-021-03819-2.
16. Mariano, D. AlphaFold e a busca pelo Santo Graal da Biologia Molecular. In: BIOINFO 02 – Revista Brasileira de Bioinformática e Biologia Computacional. Vol. 2. 2022. doi: 10.51780/978-65-992753-5-7-10
17. Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682, doi:10.1038/s41592-022-01488-1.
18. Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly Accurate Protein Structure Prediction for the Human Proteome. Nature 2021, 596, 590–596, doi:10.1038/s41586-021-03828-1.
Autores: Rafael Lemos [0000-0002-5894-2354]; Paulo Henrique dos Santos [0000-0002-3126-1751]; Aline Rocha [0000-0001-7282-957X]
Revisão: Izabela Mamede, Marcos Antonio Nobrega de Sousa, Wylerson Nogueira
Nota de transparência: este material foi originalmente produzido para um minicurso ministrado durante o Curso de Inverno em Bioinformática da UFMG, realizado em 4 de Julho de 2023, na Universidade Federal de Minas Gerais, Belo Horizonte, Brasil.
Cite este artigo:
Lemos, R; Santos, PH; Rocha, A. Extração de Informações de Sequências e Estruturas de Proteínas. BIOINFO. ISSN: 2764-8273. Vol. 3. p.12 (2023). doi: 10.51780/bioinfo-03-12
[…] Extração de Informações de Sequências e Estruturas de Proteínas […]
[…] Extração de Informações de Sequências e Estruturas de Proteínas […]