

Revisão:
BIOINFO – Revista Brasileira de Bioinformática. Edição #. .
DOI:
A Bioinformática é uma área que, em geral, trabalha com uma grande quantidade de dados, provenientes das mais diversas fontes. A Triagem Virtual se estabelece com o objetivo de selecionar as melhores moléculas a partir de diversas técnicas computacionais (in silico), visto que a pesquisa in vitro e in vivo são muito mais demoradas e dependem de mais recursos financeiros do que as técnicas in silico. Conforme a disponibilidade de dados, o pesquisador poderá realizar uma Triagem Virtual baseada no alvo, caso ele seja conhecido, ou baseado em ligante, analisando dados de uma grande quantidade de moléculas e usando técnicas de comparação entre elas, sem necessidade de usar dados do alvo. Há também a abordagem mista, quando há a junção das técnicas que analisam tanto o alvo quanto os possíveis ligantes. Por fim, aqui apresentamos uma pequena lista de ferramentas que auxiliam no processo de Triagem Virtual de ligantes.
Triagem virtual de ligantes – Uma visão geral
Na natureza, é comum que sejam encontrados dados na ordem de grandeza dos milhões ou bilhões. No processo de descoberta de fármacos (drug discovery) não é diferente. Assim, a Triagem virtual de ligantes (do inglês Virtual Screening) é indispensável para auxiliar na descoberta de medicamentos por intermédio da pesquisa de numerosas bibliotecas de pequenas moléculas. Essa técnica tem com o objetivo identificar estruturas com maior probabilidade de se ligar a um determinado alvo farmacológico, geralmente um receptor ou enzima.
Nesse contexto, duas abordagens de triagem virtual são comumente empregadas, como representado na Figura 1: as baseadas em alvos (Target-Based Virtual Screening – TBVS) e as baseadas em ligantes (Ligand Based Virtual Screening – LBVS) [1]. Na primeira, os algoritmos de triagem de ligantes, dentre um conjunto de moléculas, dependem de informações sobre os alvos para estimar as probabilidades de interação. Na segunda, não há essa dependência. No LBVS, descritores envolvendo os próprios ligantes são usados como atributos discriminantes nos algoritmos de seleção e classificação, tendo como base de conhecimento um conjunto de ligantes ativos e não ativos para determinado objetivo. Se há informação estrutural e experimental sobre o alvo, técnicas TBVS tendem a ser mais usadas, mas não necessariamente com mais sucesso [1]. Ademais, como já dito, nem sempre o alvo é bem caracterizado ou conhecido. Nesses casos, técnicas LBVS podem ser mais promissoras, ou mesmo ser a única opção.
 Figura 1: Workflow das abordagens TBVS e LBVS. Autoria própria.
Figura 1: Workflow das abordagens TBVS e LBVS. Autoria própria.
Target-based virtual screening
A abordagem Target-Based Virtual Screening (TBVS) se baseia no conhecimento da estrutura da proteína-alvo ou receptor para a execução dos processos para descoberta de ligantes.
O TBVS é interessante, especialmente, quando se tem acesso a estruturas de boa qualidade e que tenham o sítio de ligação bem caracterizado (também denominado binding site ou binding pocket) para a modelagem de um ligante no próprio sítio ativo ou a realização de um estudo do pocket para estimar, computacionalmente, a afinidade com possíveis ligantes. Tal abordagem que realiza a modelagem do ligante no pocket pode ser executada a partir de, por exemplo, complementaridade geométrica e físico-química.
Outra possibilidade, que pode atuar de forma complementar a outras técnicas, tem como principal fundamento o atracamento ou ancoramento molecular, também conhecido como docking [2]. O docking fornece novos parâmetros para a afinidade dos possíveis ligantes com a molécula-alvo [3]. Maiores detalhes a respeito do docking podem ser encontrados aqui [19].
Uma questão que vale ser destacada é que o docking, idealmente, deve ser utilizado após uma prévia seleção das moléculas a serem envolvidas. O docking é um procedimento que geralmente é mais demorado que a seleção ou filtro das moléculas envolvidas. Se for utilizado em uma abordagem em que os ligantes da molécula-alvo já sejam conhecidos, o docking pode utilizar técnicas para seleção de moléculas com características semelhantes ou diferentes às moléculas conhecidas (quando se pretende descobrir moléculas com características novas). Sabe-se que moléculas similares podem ter efeitos bioquímicos similares (MAGGIORA; SHANMUGASUNDARAM, 2011, apud [2]).
Então, se o objetivo do pesquisador for buscar moléculas comparando com os ligantes já conhecidos, a técnica TBVS deve incorporar processos inerentes às metodologias LBVS.
Ligand-based virtual screening
O Ligand-Based Virtual Screening (LBVS) tem como principal abordagem a busca de novas moléculas candidatas a partir de ligantes já conhecidos. A busca por novos ligantes pode ter como foco similaridades em suas estruturas assim como nas atividades moleculares. Segundo Cavasotto (2015), o conjunto de moléculas é dividido em dois grupos, sendo que o primeiro contém moléculas com atividade molecular altamente similar, porém, com baixa similaridade estrutural; e o segundo grupo, com baixa similaridade na atividade molecular e alta similaridade estrutural [2]. Ambas as possibilidades, se utilizados critérios bem restritos quanto ao experimento, diminuem o espaço químico de busca, uma vez que somente moléculas semelhantes serão estudadas, podendo enviesar o experimento. A próxima seção tratará das características de um experimento enviesado.
Após a busca, pode-se dar início a modelagem do possível fármaco. Para tanto, as duas etapas principais, pela abordagem LBVS, são a exploração do espaço conformacional dos ligantes e a determinação das características químicas que são comuns aos ligantes conhecidos e que são responsáveis pela ligação com a molécula-alvo (binding). Para identificar quais são essas características, geralmente, os ligantes são alinhados e comparados por tamanho, forma (shape methods), distribuição de carga e estados conformacionais [2].
A busca enviesada por moléculas
Quando o objetivo é a busca por moléculas com semelhante nível de atividade biológica dos ligantes conhecidos, encontrar moléculas que não sigam o “viés do análogo”, ou analogue bias [4] é um desafio. O “viés do análogo” é caracterizado pela busca de moléculas muito semelhantes entre si, restringindo a descoberta de plataformas de ligantes quimicamente diferentes, mas eficientes, consequentemente, reduzindo o espaço químico de análise e as possibilidades de inovação.
Uma outra possibilidade, também enviesada, é o que pode ser denominado como “viés do enriquecimento artificial” [5], que consiste em comparar um conjunto de moléculas ativas com outras muito dissimilares, podendo gerar diferenças enviesadas. Por exemplo, se um pesquisador descobre ou já conhece determinada molécula, de grande peso molecular e boa energia de ligação com o alvo pesquisado, a comparação com outras moléculas de pesos moleculares muito mais baixos poderá dar a ilusão de que ele foi um excelente candidato, com score bem destacado dos demais, quando na verdade o que de fato ocorreu é que foram feitas comparações injustas.
Também não é interessante que o conjunto de moléculas seja enriquecido artificialmente pois a avaliação de um algoritmo de pontuação ou mesmo de mineração de dados poderá tendenciar a indicação de moléculas semelhantes às ativas, o que pode não ser desejado. Há, então, de se buscar um equilíbrio entre esses dois vieses, constituindo um espaço de busca que não se limite a análogos de um dado ligante de referência, nem se pulverize entre candidatos a ligantes muito diferentes entre si.
Para que a base de dados de moléculas não seja enviesada é importante que as estruturas, pertencentes a esse conjunto, sejam obtidas de diversas fontes e que não haja uma seleção ou filtro prévios, de forma que o espaço químico seja o mais completo e diversificado possível. Porém, sabe-se que se ter um espaço químico completo, de acordo com [6], é praticamente impossível, já que o total de pequenas moléculas orgânicas que populam o “espaço químico” tem sido estimado entre 1060 [6] e 10100 [7], números muito maiores do que a quantidade de moléculas que já foram feitas e ainda serão. A título de comparação, de acordo com [7], o espaço químico de moléculas que podem ser sintetizadas é de apenas 106 e a idade do universo, estimado em segundos, é na ordem de 1017 [8].
Sendo assim, o ideal é a utilização de um conjunto de moléculas não enviesado para que, de posse de informações sobre a molécula-alvo e/ou de ligantes conhecidos, a partir de ferramentas, seja possível identificar novas moléculas a serem estudadas como prováveis ligantes. Sabe-se, no entanto, que algumas características físico-químicas precisam ser obtidas de todo o conjunto, para que sejam utilizadas como parâmetro de comparação. Essas propriedades podem ser obtidas juntamente com o arquivo da molécula (quando obtida a partir de uma biblioteca de moléculas) ou podem ser calculadas com o auxílio de um software como o RDKit [9]. A comparação das moléculas entre si também pode ser feita a partir do fingerprint de cada uma delas.
Uma vez que a abordagem LBVS tende a lidar com uma grande quantidade de moléculas, é realmente interessante avaliar se as mesmas deveriam ser agrupadas para que um estudo detalhado seja feito somente por representantes desses grupos. Detalhes sobre agrupamentos ou fingerprints serão abordados em publicações futuras.
Abordagem mista baseada no alvo e no ligante
De acordo com [10], as diversas técnicas in silico para descoberta de novos fármacos podem ser combinadas e utilizadas em conjunto. O LBVS pode ser utilizado juntamente com o TBVS, quando se tem conhecimento da molécula-alvo e de alguns ligantes dessa molécula.
Pode ser feita a classificação de moléculas em uma biblioteca de compostos de acordo com o cálculo da probabilidade de a molécula ter, conforme verificação do pesquisador, boa energia de ligação à molécula-alvo. Nesse caso, utiliza-se de um conjunto de moléculas ativas (ligantes) e outro conjunto de moléculas inativas para treinar um algoritmo classificador. Este avalia, por meio de diversos métodos de aprendizado de máquina, se uma molécula, dentro de um conjunto desconhecido, seria ativa ou inativa. Cavasotto [2] diz que esse é um método ainda pouco utilizado e não necessita de informações sobre a molécula-alvo; porém, se forem utilizadas técnicas da abordagem TBVS, a precisão dos resultados pode melhorar consideravelmente.
Avaliação de Ferramentas de Triagem Virtual
A Tabela 1 fornece um comparativo entre as ferramentas de Triagem Virtual disponíveis atualmente, ordenadas por data de publicação, ou de criação da ferramenta, da mais antiga para a mais recente. Essas ferramentas foram analisadas considerando as limitações da plataforma, tais como o limite de docking diários, detecção ou não de pockets, quantidade de moléculas disponíveis para realização do docking e outros detalhes. As conclusões obtidas são apresentadas a seguir.
Nome | Fonte targets; hits | Visualização de moléculas | Limitações |
DockingServer (2009) | Upload e RCSB PDB; PubChem | JSMol | Dois docking diários; Registro no sistema para utilização |
SwissDock (2011) | RCSB PDB; Zinc | JSMol | 264 proteínas disponíveis; não detecta pockets |
DockThor (2014) | Upload; Upload | JSMol | Não detecta pockets; Aprovação de projeto para mais de 1000 moléculas |
EasyVS (2019) | Upload e RCSB PDB; Upload, diversas bibliotecas | NGLView | Envio de pequenas moléculas somente no formato SMILES; não é possível agrupar moléculas além dos grupos pré-processados |
Alguns dos pontos que foram analisados dizem respeito a possibilidade de envio ou não de arquivos (tanto para proteína-alvo quanto para pequenas moléculas/hits), se existem bibliotecas de compostos disponíveis e, dessas, podem ser aplicados filtros para seleção de um conjunto de moléculas a partir de propriedades físico-químicas, entre outras características.
Uma das principais características verificadas, por essas serem ferramentas acessíveis pela internet, diz respeito ao design do sistema com foco na responsividade: se o sistema reage às necessidades dos usuários e seus dispositivos, alterando a disposição e/ou apresentação do conteúdo em diferentes formas e tamanhos de telas. Esse termo (design responsivo) foi primeiramente definido por [11].
DockingServer
O DockingServer (disponível em www.dockingserver.com/web) [12] é uma ferramenta que permite o docking entre proteínas e pequenas moléculas. Com a possibilidade de mais recursos a usuários que pagam pela plataforma, a ferramenta estabelece diversas limitações para usuários visitantes e que realizaram o registro gratuito na plataforma. Dentre as limitações, estão a quantidade de docking diário, espaço para armazenamento do resultado e número de processadores dedicados, dentre outros.
A referida ferramenta utiliza, para o docking, o AutoDock 4 [13]. A seleção de pequenas moléculas para docking pode ser feita somente pela busca manual (pelo nome) entre as moléculas disponíveis ou pelo upload de arquivos.
SwissDock
SwissDock (disponível em http://www.swissdock.ch) [14] é um serviço web que prediz interações moleculares que podem ocorrer entre uma proteína alvo e uma pequena molécula, podendo ser automaticamente preparadas para o docking. SwissDock utiliza o EADock DSS engine e, após testes, foi possível identificar algumas limitações, descritas a seguir.

Aqui vale salientar que no processo de testes da referida ferramenta foram feitos envios de um arquivo para target e outro como ligante. Durante a utilização da ferramenta, o ligante enviado não pode ser encontrado, porém, a interface do sistema apresentou muitas moléculas disponíveis em uma lista, mostrando que o conjunto de moléculas enviadas por usuários não registrados são compartilhadas com os demais também não registrados. Sendo assim, o teste não pode ocorrer como programado.
SwissDock possui apenas 264 alvos (no artigo e no site não há descrição dos critérios utilizados para a seleção desses alvos). Essa limitação de quantidade de alvos limita o pesquisador no estudo de ligantes para somente os alvos disponíveis na ferramenta.
Para usar a ferramenta é necessário inserir a molécula que será usada no docking através do nome descrito na base de dados ZINC, da categoria de molécula ou upload de arquivo (até 5MB). Caso o usuário queira realizar o filtro das moléculas, é possível fazê-lo pelo nome ou categoria da molécula, sem a busca por propriedades físico-químicas, que seria bastante útil.
O sistema também não detecta os pockets automaticamente, não mostra as moléculas (targets ou ligantes), não exige e-mail para recuperação dos resultados e a página web não é responsiva. SwissDock está disponível em <http://www.swissdock.ch>.
DockThor
DockThor [15] também é uma ferramenta para docking entre proteína e ligante, desenvolvida no Brasil. O sistema faz docking usando um algoritmo desenvolvido pelo próprio grupo e o JSMol para visualização dos resultados. O DockThor é hospedado no supercomputador brasileiro, chamado Santos Dumont, localizado em Petrópolis – Rio de Janeiro, disponibilizando em seu próprio portal a utilização da ferramenta através da infraestrutura de alto desempenho do SINAPAD (Sistema Nacional de Alto Desempenho). DockThor está disponível em <https://www.dockthor.lncc.br>.

Entretanto, DockThor apresenta algumas limitações como no fato de não detectar pockets, não possuir uma base de dados de proteínas e hits, além de exigir registro para dockings com mais de 1.000 moléculas (sendo necessário o envio e aprovação de um projeto).
EasyVS
O EasyVS [10, 16] é uma ferramenta que possibilita um conjunto amplo de funcionalidades, muitas delas já citadas nas demais ferramentas.
 Figura 5: Tela inicial da ferramenta EasyVS. Fonte: <http://biosig.unimelb.edu.au/easyvs/>, acesso em 6 ago. 2021.
Figura 5: Tela inicial da ferramenta EasyVS. Fonte: <http://biosig.unimelb.edu.au/easyvs/>, acesso em 6 ago. 2021.
Primeiramente, o usuário seleciona uma proteína alvo a partir do PDB ID ou faz o upload do arquivo desejado. A molécula selecionada será preparada para as demais etapas, caso a mesma já não se encontre previamente processada na base de dados interna do sistema (no caso de escolha de PDB ID, já que no upload de proteína alvo o arquivo sempre é processado). Há uma opção para o usuário inserir seu e-mail para ser notificado quando as tarefas por ele solicitadas forem finalizadas.
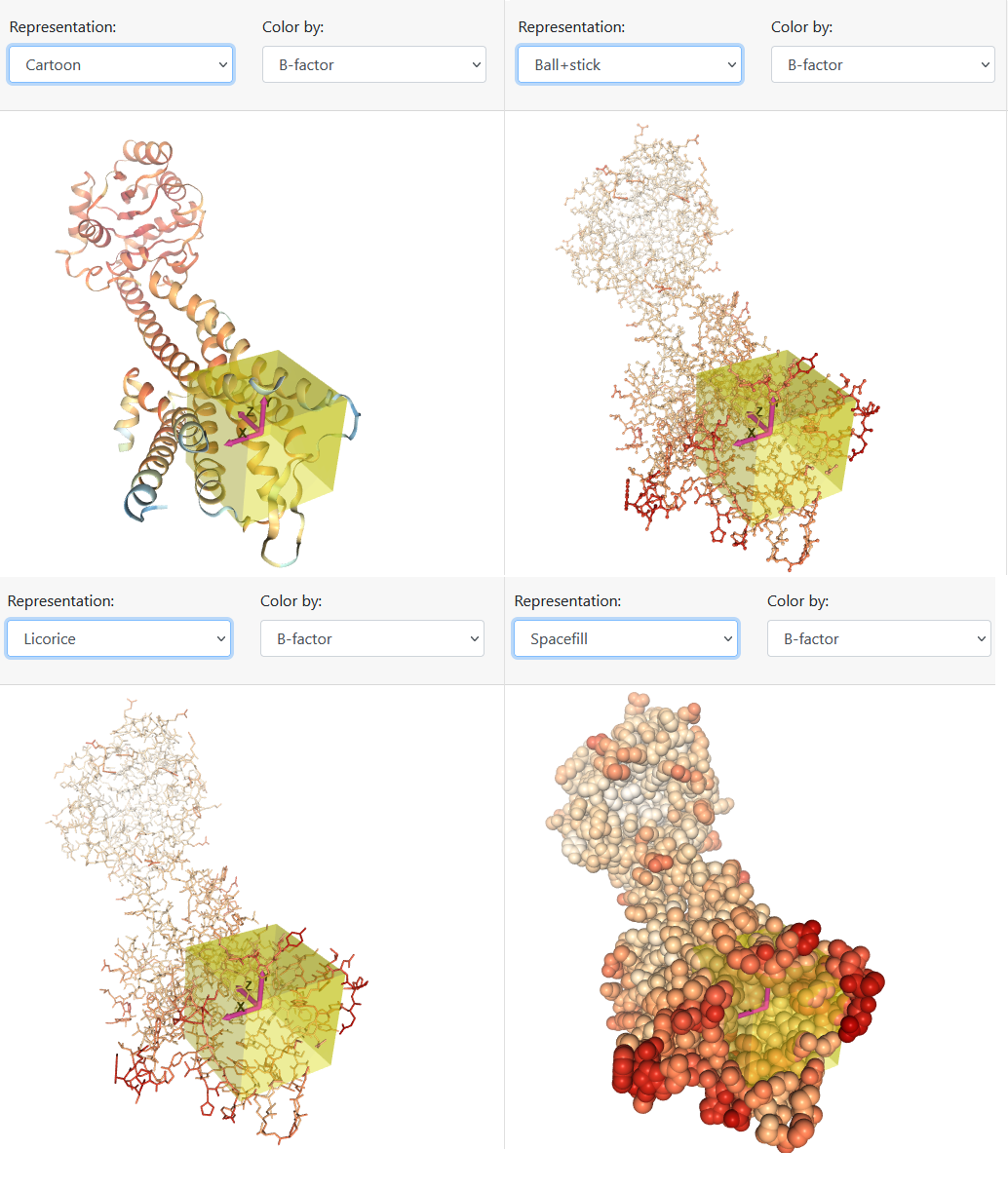
Como segunda etapa, a proteína alvo é exibida, mantendo ou não moléculas de água e heteroátomos (conforme escolhido na primeira etapa), juntamente com as configurações para realização do docking. Basicamente, o usuário visualiza a proteína a partir de cartoon, ball e sticks, licorice ou spacefill (Figura 6) e lhe é apresentado uma lista de pockets encontrados pelo Ghecom, em ordem decrescente de volume. O usuário pode selecionar algum dos pockets sugeridos ou informar coordenadas para que o centro do box seja posicionado.
Para o docking, além do posicionamento manual do centro do box, algumas configurações avançadas são permitidas. São elas: tamanho do box, exhaustiveness, número máximo de poses geradas para cada pequena molécula e energy range. Todos esses parâmetros são específicos para o Autodock Vina, software utilizado para docking no EasyVS.
Após a configuração do docking, a terceira etapa consiste na seleção de moléculas para estudo. Diferentemente de algumas das ferramentas aqui descritas, o EasyVS possui uma biblioteca de moléculas bastante variada, abrangendo DrugBank, HMDB (Human Metabolome Database), Maybridge, ChEMBL, Supernatural, Zinc e Chembridge. Caso esse conjunto de moléculas não atenda ao que o usuário deseja, pode-se fazer upload de um arquivo com SMILES de moléculas para o estudo. Essas moléculas ficarão acessíveis somente pelo pesquisador em questão.
Além da escolha de quais bibliotecas de compostos o estudo utilizará, é possível a aplicação de diversos filtros com propriedades físico-químicas das moléculas envolvidas. Pode-se filtrar as moléculas, dentre as que estão disponíveis na ferramenta, a partir de número de átomos, peso molecular, quantidade de doadores e aceptores de hidrogênio, número de anéis, ligações rotacionáveis e valor LogP.
O agrupamento das moléculas envolvidas pode ser realizado a partir da similaridade das moléculas com base em um valor de cutoff. O algoritmo utilizado no agrupamento foi elaborado por [17]. Nele, há a garantia que a similaridade selecionada pelo usuário seja respeitada, tendo como entrada para o algoritmo os fingerprints das moléculas selecionadas, usando o MACCS166. A título de exemplo, caso a similaridade escolhida seja 0.85, essa opção fará com que as moléculas selecionadas sejam agrupadas em grupos com similaridade de, ao menos, 85% dos features de cada molécula com um determinado representante do grupo, denominado de centroide. Em cada grupo, uma molécula é selecionada aleatoriamente para que seja feito o docking e o resultado seja apresentado na etapa quatro.
Finalmente, na última etapa (Figura 7), todas as moléculas são apresentadas, conforme o resultado do docking for sendo disponibilizado pela ferramenta. Salienta-se que há uma previsão de tempo para que o processamento seja finalizado (tempo este apresentado ao usuário na etapa 3). Porém, o tempo prevê somente o processamento do conjunto de moléculas selecionadas em si, e não os processos de outros usuários que possam estar sendo executados. Para cada resultado apresentado, é possível visualizar a afinidade entre a pequena molécula e o alvo, as poses, propriedades físico-químicas, além da possibilidade de download de todas as poses preditas (Figura 8), o estudo da molécula em si pelo pkCSM [18] e a otimização da molécula usando o próprio EasyVS (Figura 9).
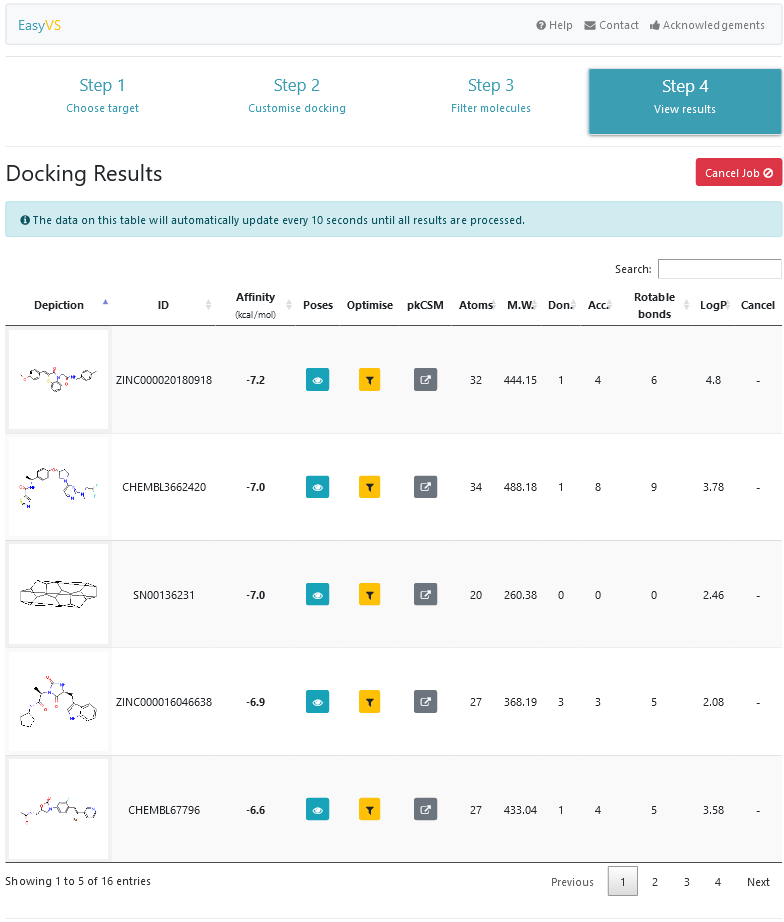

A otimização do resultado apresentado pela ferramenta (Figura 8) consiste em criar um novo espaço químico conforme a molécula selecionada. Para isso, o usuário visualiza uma determinada molécula que tiver um resultado que considerar interessante, clica no botão “Optimise” e seleciona o nível de similaridade com a molécula em questão. Essa opção irá retornar a etapa anterior (etapa 3) com um conjunto de moléculas semelhantes à molécula de referência. Esse processo poderá ser repetido indefinidamente, o que possibilita que o usuário encontre resultados com afinidades, entre as moléculas envolvidas, cada vez maiores.

Por fim, cabe ressaltar que maior parte das ferramentas analisadas estabeleciam limitações de recursos ou de funcionalidades para seus utilizadores, podendo ser devido à grande utilização e objetivando prover o acesso a uma maior quantidade de pesquisadores. A única ferramenta analisada que ainda não limita recursos a seus utilizadores é o EasyVS, porém, sabe-se que o tempo de resposta para obtenção de resultados pode variar conforme a quantidade de requisições ao servidor e, devido a essa questão, limitações semelhantes às demais ferramentas podem ser estabelecidas.
Referências
[1] Domingues & Lopes, 2012 – Domingues, B. F.; Lopes, J. C. D. (2012). 3D-Pharma: Uma Ferramenta para Triagem Virtual Baseada em Fingerprints de Farmacoforos. http://www.bibliotecadigital.ufmg.br/dspace/handle/1843/BUBD-9DKHDA
[2] CAVASOTTO, Claudio N. (Ed.). In silico drug discovery and design: theory, methods, challenges, and applications. CRC Press, 2015.
[3] VERLI, Hugo. Bioinformática: da biologia à flexibilidade molecular. 2014.
[4] GOOD, Andrew C.; OPREA, Tudor I. Optimization of CAMD techniques 3. Virtual screening enrichment studies: a help or hindrance in tool selection?. Journal of computer-aided molecular design, v. 22, n. 3, p. 169-178, 2008.
[5] VERDONK, Marcel L. et al. Virtual screening using protein− ligand docking: avoiding artificial enrichment. Journal of chemical information and computer sciences, v. 44, n. 3, p. 793-806, 2004.
[6] Kirkpatrick, P. & Ellis, C. (2004). Chemical space. Nature, 432:823–823. ISSN 0028- 0836.
[7] WALTERS, W. Patrick; STAHL, Matthew T.; MURCKO, Mark A. Virtual screening-an overview. Drug discovery today, v. 3, n. 4, p. 160-178, 1998.
[8] ADE, Peter AR et al. Planck 2015 results-xiii. cosmological parameters. Astronomy & Astrophysics, v. 594, p. A13, 2016.
[9] Landrum, G. (2006). RDKit Documentation.
[10] VELOSO, W. N. P. Easyvs: uma ferramenta para triagem virtual mista baseada em alvo e ligante. Tese de Doutorado – Universidade Federal de Minas Gerais. Belo Horizonte. 2019. Disponível em: <http://hdl.handle.net/1843/30754>, acesso em 21 maio 2021.
[11] Marcotte [2010] – Marcotte, E. (2010). Responsive Web Design. http://alistapart.com/article/responsive-web-design
[12] BIKADI, Zsolt; HAZAI, Eszter. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. Journal of Cheminformatics, v. 1, n. 1, p. 1-16, 2009.
[13] Morris et al., 2009 – Morris, G. M., Ruth, H., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., & Olson, A. J. (2009). Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30(16), 2785–2791. https://doi.org/10.1002/jcc.21256
[14] Grosdidier et al., 2011 – Grosdidier, A., Zoete, V., & Michielin, O. (2011). SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Research, 39(Web Server issue), W270-7. https://doi.org/10.1093/nar/gkr366
[15] de Magalhães et al., 2014 – de Magalhães, C. S., Almeida, D. M., Barbosa, H. J. C., & Dardenne, L. E. (2014). A dynamic niching genetic algorithm strategy for docking highly flexible ligands. Information Sciences, 289, 206–224. https://doi.org/10.1016/J.INS.2014.08.002
[16] Pires, Douglas E. V., et al. “EasyVS: A User-Friendly Web-Based Tool for Molecule Library Selection and Structure-Based Virtual Screening”. Bioinformatics, vol. 36, n. 14, julho de 2020, p. 4200–02. https://doi.org/10.1093/bioinformatics/btaa480.
[17] Butina, D. (1999). Unsupervised data base clustering based on daylight’s fingerprint
and Tanimoto similarity: A fast and automated way to cluster small and large data
sets. Journal of Chemical Information and Computer Sciences, 39(4):747–750. ISSN
00952338.
[18] Pires, 2015 – Pires, D. E. V., Blundell, T. L., & Ascher, D. B. (2015). pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. Journal of Medicinal Chemistry, 58(9), 4066–4072. https://doi.org/10.1021/acs.jmedchem.5b00104
[19] Santos, LH. Docagem molecular: em busca do encaixe perfeito e acessível. In: BIOINFO – Revista Brasileira de Bioinformática e Biologia Computacional. 1. Ed. Vol. 1. Lagoa Santa: Editora Alfahelix, 2021. DOI: 10.51780/978-6-599-275326