





Este artigo tem por objetivo fornecer um entendimento histórico acerca da biologia de sistemas, da mineração de texto, da curadoria manual, das redes biológicas, e, principalmente, das redes de interação proteína-proteína (redes PPIs).
Ademais, este trabalho ainda busca traçar algumas bases para o desenvolvimento da construção das redes PPIs em diversos trabalhos acadêmicos (em variadas linhas de investigação), buscando o desenvolvimento de estudos computacionais (in silico) para um avanço sobre elucidações moleculares e celulares no que tange ao funcionamento proteico em inúmeras doenças, estados fisiológicos saudáveis e dentre outros focos de pesquisa.
Assim, este tutorial está dividido em quatro partes:
- Apresentação e explicação do tema: contexto histórico da Biologia de Sistemas, contexto histórico das Redes Biológicas, introdução e explicação sobre os componentes-chave das Redes de Interação Proteína-Proteína e Mineração de Texto;
- Desenvolvimento do tutorial: exposição dos Componentes Necessários, da Busca de Artigos, da Seleção de Proteínas e da Construção da Rede;
- Exposição conceitual sobre os dados obtidos: características da rede PPI, os Parâmetros Topológicos avaliados na rede PPI e a Avaliação Funcional da rede PPI;
- Fornecimento de Literatura Complementar: visa à divulgação de outros materiais formativos úteis ao desenvolvimento do leitor;
Matheus Correia Casottiǂ*, Débora Dummer Meira*, Carla Carvalho de Aguiar, Daniel de Almeida Duque, Kymberlin Costa de Souza, Letícia Oliveira Portella, Raquel Furlani Rocon Braga, Iúri Drumond Louro
*Contribuíram igualmente para a realização deste trabalho. ǂAutor correspondente: matheuscasotti@yahoo.com. Revisão: Diego Mariano
Organização geral
Parte 1: O que será aprendido:
- Contexto histórico da Biologia de Sistemas;
- Contexto histórico das Redes Biológicas;
- Introdução e Explicação sobre os componentes-chave das Redes de Interação Proteína-Proteína (rede PPI – protein-protein interaction);
- Mineração de Texto.
Parte 2: Mãos à obra:
- Componentes Necessários;
- Busca de Artigos;
- Seleção de Proteínas;
- Construção da Rede.
Parte 3: Entendendo a fundo:
- Características e Parâmetros Topológicos da rede PPI;
- ClusterMaker;
- Avaliação Funcional da rede PPI.
Parte 4: Sugestões de literatura complementar para aprimorar o estudo em biologia de sistemas e redes biológicas.
Parte 1 – Introdução
Contexto histórico da Biologia de Sistemas
É muito comum destacar que os sistemas biológicos, como as células, são ‘sistemas complexos’. Uma noção popular de sistemas complexos é a de um grande número de elementos simples e idênticos interagindo para produzir comportamentos “complexos”. Porém, para os sistemas biológicos, encontra-se um grande número de conjuntos de elementos funcionalmente diversos e, frequentemente, multifuncionais que interagem de maneira seletiva e dinâmica para gerar comportamentos coerentes em vez de complexos. Assim, compreender sistemas biológicos complexos requer a união entre a pesquisa experimental e computacional, ou seja, uma abordagem de biologia de sistemas[1].
A biologia de sistemas possibilitou alcançar uma estrutura para montar modelos de sistemas biológicos por meio de medições sistematizadas. Logo, ela proporciona um avanço elevado em diversos campos, como aplicações sobre: (a) biomarcadores baseados em vias, (b) mapas de interação genética global, (c) abordagens de sistemas para identificar genes relacionados a doenças e (d) biologia de sistemas de células-tronco. Além disso, o desenvolvimento científico nessa área acaba induzindo uma produção elevada de novos aplicativos e softwares que possam suprir a necessidade dos pesquisadores em realizar as análises sistêmicas de uma forma mais acessível e de fácil compreensão mesmo perante a complexidade e o abstracionismo intrínsecos[2].
Para além, como uma resultante da necessidade de se entender a biologia no nível do sistema, precisa-se examinar a estrutura e a dinâmica da função celular e do organismo, em vez das características de partes isoladas de uma célula ou organismo. As propriedades dos sistemas, como a robustez, surgem como questões centrais e a compreensão dessas propriedades pode ter um grande impacto no futuro da medicina. Outrossim, programas como Cytoscape (<https://cytoscape.org/>) demonstram o avanço da Biologia de Sistemas sobre o desenvolvimento de novos dispositivos experimentais, softwares avançados e métodos analíticos com grande potencial para o avanço da compreensão dos sistemas biológicos como um todo, associando com dados ômicos, de ontologia genética, dentre outros[3].
Contexto histórico das Redes Biológicas
Um grande conhecimento foi proporcionado pelo reducionismo, que permitiu ter um entendimento dos componentes celulares individuais e suas funções. Mas mesmo com grande sucesso, tornou-se cada vez mais claro que a função biológica não estava só relacionada com uma única molécula. Ou seja, a maioria das características biológicas surge de interações complexas, e isto se torna um desafio associado à biologia de sistemas capaz de ser avançado por meio das redes biológicas. E tal abordagem proporcionou o entendimento de que as redes celulares seguem por leis universais e oferecem uma nova estrutura conceitual que pode revolucionar a visão da biologia e das patologias das doenças[4].
Além disso, pela natureza das interações, as redes podem ser direcionadas ou não. Em acréscimo, as redes seguem propriedades e princípios simples das características das redes, como sua característica ‘democrática’ ou uniforme, caracterizando o grau, ou conectividade, a ausência de escala, ou seja, a presença de um número desproporcional de nós altamente conectados, uma característica comum de que as redes possuem quaisquer dois nós conectados com um caminho de apenas algumas bordas4.
Há também a origem evolutiva das redes, que provém de um processo de crescimento, onde novos nós se juntam ao sistema por um longo período de tempo, seguindo uma conexão preferencial, ou seja, os nós preferem se conectar a nós que já possuem muitas bordas (links/arestas), e com esse conceito, pode-se sustentar-se pelo mecanismo biológico da duplicação de genes4.
Ademais, é demonstrado também que as funções celulares seguem uma maneira modular, ou seja, sendo um grupo de moléculas fisicamente ou funcionalmente ligadas que, em conjunto, trabalham para alcançar uma função (relativamente) distinta. E em uma associação, esse módulo (ou cluster) aparece como um grupo de nós altamente interconectado4.
Com uma complementaridade das análises, o alto agrupamento indica que as redes também são ricas em pequenos conteúdos localmente com vários sub-grafos dos grupos de nós altamente interligados, o que é uma condição para o surgimento de módulos funcionais isolados. E tais sub-grafos (motivos) capturam padrões específicos de interconexões que caracterizam uma determinada rede no nível local4.
E, ao finalizar sobre uma modularidade hierárquica, indica que os módulos não têm um tamanho característico. Outrossim, destaca-se que os sistemas biológicos seguem uma robustez, ou seja capacidade de responder a mudanças nas condições externas ou na organização interna, mantendo um comportamento relativamente normal4.
E, por fim, ao destacar a aplicação prática, como por exemplo, ao se buscar um alvo mais eficaz em doenças complexas, tornou-se importante repensar as estratégias para o desenvolvimento de medicamentos e a seleção de alvos moleculares para tratamentos farmacológicos junto aos estudos das redes. Para tanto, a arquitetura de redes fornece a possibilidade da realização de medições quantitativas por meio de diversos algoritmos diferentes, visando prever as trajetórias tomadas por um sistema celular no tempo ou através dos estados de doenças, formalizando assim o campo da medicina de rede, como uma área atual e de grande impacto para estudos personalizados e complexos[5].
Introdução e explicação sobre os componentes-chave das Redes de Interação Proteína-Proteína
As redes PPIs fornecem informações valiosas para a compreensão da função celular e dos processos biológicos, isto porque, com os avanços no conhecimento das interações proteína-proteína (PPIs), novos mecanismos moleculares de doenças puderam ser compreendidos. Além disso, novos insights sobre características topológicas distintas das proteínas e seus respectivos genes puderam ser obtidos, o que permitiu melhores elucidações acerca de sub-redes relacionadas às doenças6.
Diante disto, a representação de rede abstrata, onde as proteínas são nós e as interações são bordas, algumas proteínas agem como proteínas centrais, altamente conectadas a outras, também chamadas de proteínas hub, enquanto outras possuem poucas interações. E é por meio da disfunção de algumas interações que novos insights são elucidados ou explicitados[6]. Compreende-se que as interações proteína-proteína (PPIs) são cruciais para todos os processos biológicos, logo, a compilação de redes PPI fornece novas percepções sobre a função das proteínas, os princípios de organização genérica das redes celulares funcionais e mapas de interação proteína-proteína como uma estrutura valiosa para uma melhor compreensão da organização funcional do proteoma[7].
Frente aos benefícios das redes PPIs supracitados, o entendimento dessas interações moleculares, tornam-se críticas para coordenar os mecanismos subcelulares. Isto porque tais eventos são altamente regulados dentro de uma paisagem interconectada dinâmica de vias moleculares. E, assim, comprometer tais vias moleculares pode contribuir para a sinalização aberrante e, em última instância, para a disfunção celular. Logo, as PPIs nessa paisagem subcelular, destacam-se como uma teia de “nós” (proteínas) conectados por “bordas” que representam interações. E por meio da teoria dos grafos, há a possibilidade de avaliar a interconectividade das proteínas que sustentam a funcionalidade do sistema[8].
Ademais, com a análise topológica, as interações podem destacar as proteínas hub e clusters (agrupamentos ou módulos) de proteínas. Os hubs são as peças que sustentam a conectividade da rede e os clusters funcionam como comunidades de nós com um maior grau de interconexão em relação a toda a rede, podendo representar módulos funcionais específicos e/ou complexos de proteínas8. Pode-se exemplificar hub e clusters por meio da figura 1, que se segue, onde como hub tem o nó “C” e cluster o conjunto de nós “C”, “E” e “F”.
E em consonância a isso, além de uma visão mais matemática, abarca-se também nas análises, o insight funcional em redes de interação de proteínas, em sua maioria obtido por análises de enriquecimento funcional e anotação de vias, utilizando-se de dados de projetos de anotação funcional em grande escala, como Gene Ontology (GO), Reactome, KEGG e WikiPathways, para identificar processos biológicos e vias que são particularmente relevantes para redes e/ou sub-redes PPI específicas8.


Outrossim, com base no princípio de culpa por associação, os dados de anotação funcional são úteis para inferir associações funcionais de proteínas que estão mal caracterizadas ou têm funções desconhecidas8.
Mineração de Texto
A literatura biomédica pode ser observada como um grande repositório de dados integrados, mas sem uma boa estruturação. Logo, a extração de informações da literatura, visando torná-las mais acessíveis, segue uma linha de curadoria manual, na qual busca criar ontologias e vocabulários para anotar produtos genéticos com base em declarações em artigos. E em uma linha, tem-se a mineração de texto que busca identificar automaticamente essas entidades e seus relacionamentos no texto usando técnicas de recuperação de informações e processamento de linguagem natural. Isto porque a curadoria manual é altamente precisa, mas demorada, e se torna incompatível com a elevada quantidade crescente de dados da literatura. Enquanto isso, a mineração de texto como uma técnica computacional de alto rendimento é bem escalável, mas passa a ficar aberta a erros devido à complexidade da linguagem natural do próprio pesquisador[9].
Então, em resumo, a curadoria manual de alta qualidade da literatura científica é um gargalo comum para o preenchimento de bancos de dados biológicos, pois se torna desafiador acompanhar o crescente volume e escopo dos dados biológicos publicados[10]. Logo, a curadoria manual encontra três problemas principais: (i) escalabilidade, ou seja, devido à alta quantidade de dados e literaturas, a curadoria manual não consegue acompanhar; (ii) evolução, ou seja, as ontologias usadas acabam mudando ao longo do tempo por meio da adição, substituição ou movimentação de conceitos; e (iii) acordo entre anotadores, ou seja, a anotação perpassa pela subjetividade, dependendo da formação científica e a experiência do próprio pesquisador9.
Diante do que foi explicitado, o impacto e a importância da mineração de texto vêm aumentando sobre o gerenciamento de serviços à medida que o acesso à “big data” cresce nas plataformas digitais[11]. Dessa maneira, a mineração de texto biomédica possibilita gerenciar a sobrecarga de informações, solucionar problemas específicos, recuperar documentos relevantes ou extrair pedaços de informações de documentos, usar técnicas para recuperação de informações, extração de informações, classificação de texto e métodos de aproveitamento de campos relacionados[12].
Ademais, os avanços atuais na mineração de texto alcançam curadores de bancos de dados de organismos modelo e cientistas de bancada sobre a construção de aplicativos de mineração de texto, possibilitando lidar com questões diversas da literatura científica com publicações relacionadas às ômicas[13]. Porém, não se pode excluir os benefícios proporcionados pela curadoria manual como uma fonte primária de dados para a formulação e organização de trabalhos e pesquisas sustentados em uma abordagem muito robusta que justifica a subjetividade aplicada.
Parte 2 – Componentes Necessários
- Computador com acesso à internet;
- Download do Cytoscape: Acesse a página do Cytoscape em <https://cytoscape.org/> e baixe o programa na aba Download/Baixar que aparece em destaque na cor laranja (no software atual de 2021).
- Download dos aplicativos no Cytoscape:
- Apps ->App manager -> caixa de texto Search -> escreva stringApp -> clique em install;
- Apps ->App manager -> caixa de texto Search -> escreva NetworkAnalyzer -> clique em install;
- Apps ->App manager -> caixa de texto Search -> escreva cytoHubba -> clique em install;
- Apps ->App manager -> caixa de texto Search -> escreva clusterMaker2 -> clique em install;
- Feche App manager -> símbolo “X” ou clicando em close.
- Seleção de palavras-chave, previamente, ou seja, selecionar palavras ou termos biológicos de interesse para pesquisar artigos. Por exemplo: cancer, cancer AND genome, cancer OR genome, e etc.
Busca de Artigos
Um dos maiores sites de pesquisa é o PubMed que é inteiramente gratuito e compreende periódicos científicos, livros on-line e literatura biomédica. É possível acessá-lo através de qualquer dispositivo conectado à internet (como computador, tablet e celular). E por meio dele é possível alcançar a pesquisa de diversos artigos científicos em diversos impactos diferentes. Por exemplo, vamos realizar a seleção de proteínas, a partir de artigos, para a construção de uma rede.
Passo 1: para fazer uma busca simples sobre o tema do seu interesse no PubMed, basta acessar o endereço https://pubmed.ncbi.nlm.nih.gov/ e você será direcionado para a página inicial do site, como exemplificado abaixo:


Atenção! Não há necessidade de realizar login. Basta digitar na aba de pesquisa as palavras-chave desejadas e clicar no botão verde Search ou pressione a tecla Enter.
Passo 2: após efetuar a pesquisa, uma tela semelhante a imagem abaixo irá aparecer:


Atenção! Irá aparecer uma lista de artigos científicos relacionados ao termo que você pesquisou. A maioria dos artigos científicos é escrito em inglês, pois a literatura científica tem a língua inglesa como a padrão.
Passo 3: após encontrar o artigo de seu interesse, clique no título do artigo, como exemplificado abaixo (em roxo):


Passo 4: ao clicar no título do artigo escolhido, você será direcionado para outra página que na maioria das vezes contém apenas o resumo (abstract) do artigo. Para ter acesso ao artigo completo, clique no número em azul, chamado de DOI, que significa Digital Object Identifier (Identificador de Objeto Digital), sendo um padrão de números e letras que identificam publicações:

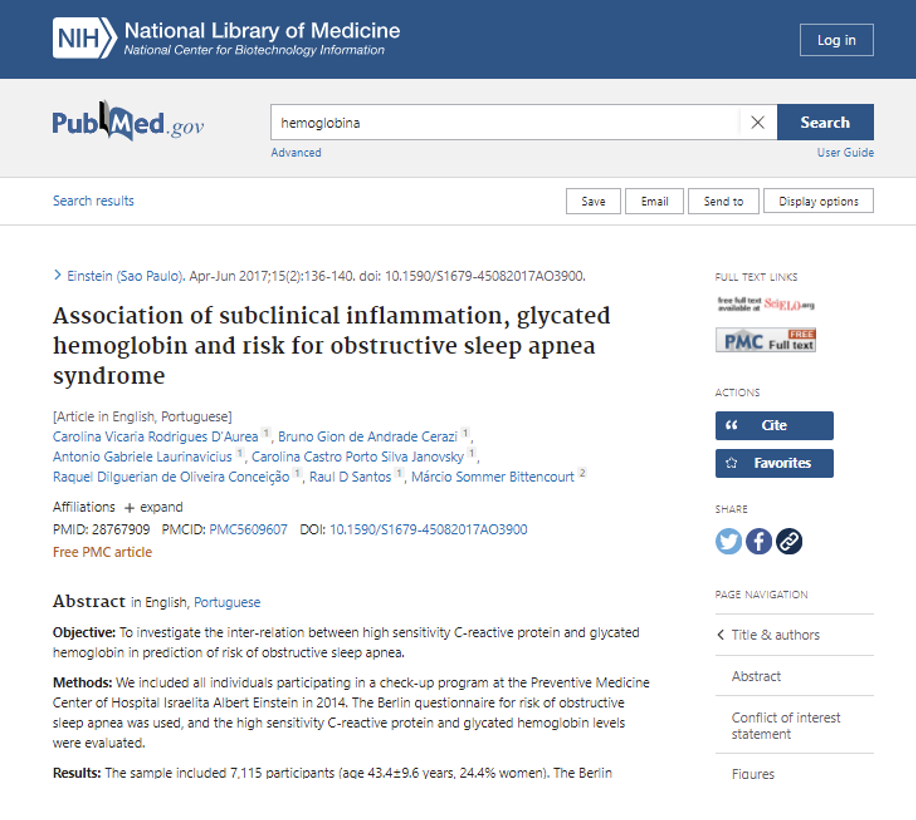
Passo 5: após clicar no DOI, você será direcionado, mais uma vez, para outra página e nela você terá acesso ao artigo:


Atenção! Segue um fluxograma resumindo a etapa inicial referente à Curadoria Manual:


Seleção de Proteínas
Após a busca dos artigos, é preciso ser feita a leitura dos materiais incluídos no estudo segundo a figura 1, para então selecionar as proteínas, para que possa ser feita a procura dos códigos das proteínas obtidas.
Passo 1: Proteínas selecionadas, podem ser direcionadas ao Uniprot para pesquisar os códigos associados a elas. Para isto, basta acessar<https://www.uniprot.org/> e escrever na caixa superior de pesquisa o nome das suas proteínas (apontado pela seta vermelha). Depois, siga para a aba inferior onde está escrito “Protein names/Nomes das proteínas” e copie o nome de cada uma das proteínas, previamente selecionadas para a busca, INDIVIDUALMENTE, como se segue:


Atenção! Selecione o organismo analisado por você, por exemplo: Homo sapiens (Human).
Passo 2: Com a pesquisa INDIVIDUAL de cada proteína, como por exemplo no caso da pesquisa do antígeno de p53, como segue no exemplo abaixo, pode-se buscar o código de acesso (apontado pela seta em vermelho):


Construção da Rede
O Cytoscape é uma das ferramentas para exploração visual de redes biomédicas compostas de proteínas, genes e outros tipos de interações. Esse software fornece funcionalidades básicas para carregar, visualizar, pesquisar, filtrar e salvar redes e centenas de aplicativos e estendem essa funcionalidade para atender a requisitos específicos necessários à pesquisa. Para isso, há um grande número de aplicativos (também conhecidos como plugins) projetados para a análise de redes biológicas, permitindo a importação de dados de repositórios externos, anotação funcional e descoberta, detecção de módulo, pesquisa de literatura, layouts e filtros de redes.
Antes de começar a construção da rede, é importante instalar o aplicativo stringApp no Cytoscape. Em seguida, deve-se direcionar às próximas etapas para construção da rede de interação proteína-proteína. Para isso, basta seguir os passos a seguir:
Passo 1: Clique emApps ->App Manager.


Passo 2: A seguir, clique em Search->e digite “stringApp” -> e clique em “Install”.


Passo 3: Utilize uma lista de proteínas/genes selecionados com base em curadoria manual e seleção de proteínas. Para esse passo, siga para https://www.uniprot.org/, pesquise cada proteína e anote em uma tabela o código do Uniprot. Por exemplo:


Passo 4: Após a organização e a digitação dos códigos do Uniprot. Copie o código Uniprot (entry), abra o Cytoscape e escolha STRING protein query. Como a seguir:


Passo 5: Após copiar e colar os códigos como no passo 4, direciona e clique nas três barras à direita, circundado em vermelho, como se segue.


Passo 6: E, após a seleção das três barras (circundado em vermelho) selecione a espécie analisada, como Homo sapiens. Em seguida, selecione a rede completa (full STRING network), e ao final padronize os parâmetros do limiar da pontuação de confiança, confidence (score) cutoff, equivalente a 0.70, ou seja, 70%. A seguir, selecione como um máximo de interatores (maximum additional interactors), o valor 100, e ao final clique no símbolo da lupa e espere carregar a formação da rede, como em destaque:


Passo 7: Com a rede montada, é necessário realizar uma seleção de rede para analisar o aspecto topológico. Siga para ferramentas (tools), e selecione a opção Analyze Network (analisar rede):


Passo 8: clique em Style (aba à esquerda; seta vermelha), selecione a opção STRING style v1.5:


Depois marque a opção do estilo default, como segue na imagem a seguir:


Passo 9: Agora, realize a configuração das propriedades do estilo (style) referentes às cores e tamanhos de bordas em relação à centralidade de intermediação (Betweenness Centrality), assim como a coloração do nó em relação ao grau (Degree), obtido pelo Analyzer Network. A cor de borda (Border paint) pode ser configurada desta forma:


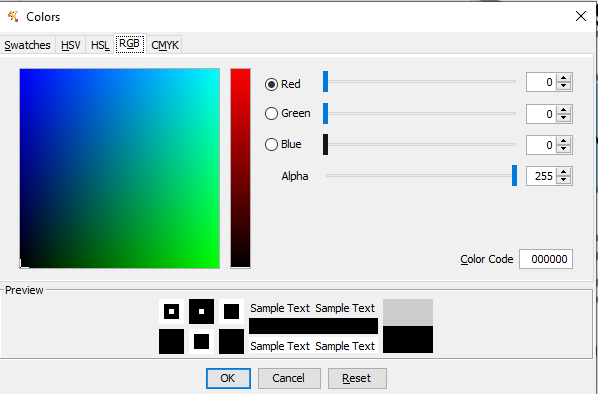
Para escolher uma cor escura, clique em Def. -> RGB -> e preencha todo o campo color code com 0:

Para configurar espessura ou tamanho da Borda (Border Width), vá em: Column: Betweenness Centrality e Mapping Type: Continuous Mapping. Em seguida, clique no gráfico para configurar o tamanho mínimo e máximo da borda:


Para definir a cor de preenchimento de cada Nó (Fill color), vá em: Column: Degreee Mapping Type: Continuous Mapping. E, em seguida, clique no gráfico como ilustra a imagem a seguir:


Depois, selecione do valor mínimo (0) ao máximo, que no exemplo foi usado do branco (mínimo) ao vermelho (máximo), como na figura abaixo:


Para configurar o rótulo, ou seja, o nome que será exposto no Nó (Label), vá em Column: display name, desta forma:


Passo 10: Após a avaliação dos parâmetros topológicos da rede, você pode avançar para uma análise modular e funcional da rede. Esta última etapa é crucial para as sugestões de alvos candidatos como biomarcadores, para sugerir alvos de fármacos, para elucidar de mecanismos moleculares e celulares pouco compreendidos, dentre outras aplicações proporcionadas pelas redes PPIs. Assim, a seguir, será direcionado para a modularização da rede por meio do uso do clusterMaker e o algoritmo de clusterização MCODE, como demonstra a figura a seguir:


A seguir, selecione a opção fluff e depois o tópico create new clustered network:


Após a realização das etapas atreladas às imagens acima, observar os módulos (clusters ou complexos) formados:


Passo 11: Após a obtenção dos módulos, selecione novamente a rede principal e vá para a aba apps, e em seguida basta selecionar o aplicativo do cytoHubba, como destaca a figura a seguir:


Em seguida, aparecerá uma janela com cinco caixas de texto (“Target Network”, “Nodes’Scores”, “Select nodes”, “Particular nodes” e “Display options”). Na caixa de texto “Target Network” aparecerá o nome da rede. A seguir, em “Nodes’ Scores” clique em “Calculate”. Em “Select nodes”, selecione a opção “Top” e digite 10 na caixa ao lado. Na aba inferior, selecione “BottleNeck” e, ao final, clique em “Submit”.

Passo 12: Após a finalização de todas as análises provindas de aplicativos específicos, pode-se realizar a análise funcional da rede principal, dos módulos e dos candidatos selecionados pelo cytoHubba através da seleção da rede principal e sub-redes geradas pelos programas. Para isso, escolha a opção STRING Enrichment -> Retrieve functional enrichment:


Após a seleção do Retrieve functional enrichment, aparecerá a seguinte caixa de texto:


Clique em OK.
Ao final, aparecerá na tabela dos nós e das arestas (inferior) uma aba onde poderá ser encontrada uma lista de relações funcionais segundo diversas bases de dados, como destaca a imagem a seguir:


Caso for de interesse filtrar de acordo com alguma ontologia genética ou a alguma base de dados, basta clicar no ícone de funil (apontado pela seta vermelha), selecionar o filtro desejado e clicar em OK, como está na imagem a seguir:


Passo 13: Com a finalização de todas as análises, pode-se exportar a rede principal e as sub-redes obtidas no estudo realizado, por meio da organização a seguir:


Também se pode exportar como tabela:


Atenção! Segue um fluxograma resumindo a etapa referente estudo prático in silico:


Parte 3 – Características e Parâmetros Topológicos da rede PPI
Com a análise inicial por meio do Network Analyzer, a rede PPI gerada por meio das proteínas obtidas irá fornecer o cálculo de parâmetros topológicos que demonstraram, como por exemplo:
- (A) Grau: Número de conexões realizadas por um nó focal. Ou seja, grau do nó “C” = 3 (realiza três conexões). Exemplo de rede não direcionada:


- (B) Distribuições de graus: fornece o número de nós com respectivo grau. Ou seja, quantos nós possuem uma conexão, duas conexões, e assim por diante. Exemplo da distribuição de nós em uma rede biológica real:
Figura 4: Distribuição do Grau dos Nós.


Fonte: Próprio Autor.
- (C) Comprimento do caminho mais curto: menor caminho para percorrer a rede de um nó “A” a um nó “G”. Logo, sendo o número de conexões que o formam. Quando evidencia um comprimento médio do caminho mais curto, refere-se ao comprimento do caminho característico que fornece a distância esperada entre dois nós conectados.
Ou seja, nesse exemplo abaixo, para ir do nó “A” ao nó “G”, pode seguir por A -> D -> B -> C -> E -> F -> G ou A -> D -> B -> C -> F -> G (Menor caminho de conexão entre o nó “A” ao nó “G”.


- (D) Diâmetro da rede: comprimento máximo dos caminhos mais curtos entre dois nós. E indica propriedades da característica do mundo pequeno.
- (E) Coeficientes de agrupamento: número de triângulos (três loops) que passam por esse nó, representando uma razão do número de arestas entre os vizinhos dos nós / número máximo de arestas que podem existir entre os nós, em relação ao número máximo de três loops que podem passar pelo nó, sendo sempre um número entre 0 e 1. Assim, quanto maior a força na criação de grupos unidos por uma densidade relativamente alta de laços, maior é o coeficiente de agrupamento. Logo, pode-se sugerir um alto coeficiente para o nó “C”, sendo necessária a comprovação matemática:


- (F) Coeficientes topológicos: medida relativa para a extensão em que um nó compartilha vizinhos com outros nós. Ou seja, é uma medida para determinar até que ponto um nó compartilha vizinhos com outros nós.
- (G) Centralidade de intermediação: quantidade de controle que este nó (observado na análise) exerce sobre as interações de outros nós na rede e sempre favorece nós que se unem a comunidades/módulos (sub-redes densas). Então, com base nessa definição, pode-se perceber que o nó “C” representa um nó onde há uma maior passagem de informação do que em relação aos demais nós, como se observa a seguir:


- (H) Centralidade de proximidade: medida de quão rápido a informação se espalha de um determinado nó para outros nós. Ou seja, o nó mais central, estaria sendo aquele mesmo nó mais próximo da maior parte de todos os demais nós. Para tanto, a seguir pode ser exemplificado o papel do nó “C” como um nó com alta centralidade de proximidade:


ClusterMaker
No aplicativo clusterMaker foi utilizado como algoritmo o MCODE, o qual encontra regiões altamente interconectadas em uma rede. Para isso, ele usa um processo de três estágios:
- Ponderação de vértice: a qual é feita uma ponderação de todos os nós com base na densidade da rede local;
- Predição do complexo molecular: que começa com o nó de maior peso, move-se continuamente adicionando nós ao complexo que estão acima de um determinado limite;
- Pós-processamento: que aplica filtros para melhorar a qualidade do cluster, módulo ou agrupamento.
Além disso, o MCODE fornece alguns ajustes avançados para a pontuação da rede, como: Incluir loops, loops (arestas próprias) são incluídos no cálculo para a ponderação do vértice; e Corte de grau, controla o grau mínimo necessário para que um nó seja pontuado.
Ajustes avançados também podem ser feitos sobre a busca dos clusters, módulos ou agrupamentos, por meio dos seguintes parâmetros:
- Haircut: elimina todos os nós de um cluster se eles possuírem apenas uma única conexão com o cluster;
- Fluff: após o haircut, todos os núcleos do cluster são expandidos em uma etapa e adicionados ao cluster se a pontuação for maior que o corte de pontuação do nó;
- K-Core: filtra os clusters que não formam um subconjunto maximamente interconectado com um dado conjunto mínimo de graus;
- Profundidade Máxima: controla o quão longe do nó semente (o objeto de estudo – genes, proteína ou outro – inserido pelo pesquisador) o algoritmo irá pesquisar na etapa de predição do complexo molecular.
Avaliação Funcional da rede PPI
Para a avaliação funcional pode-se utilizar comandos por meio do stringApp, que permite a alteração da pontuação de confiança e expansão da rede por um número arbitrário de nós. Pode-se usar proteínas do mesmo organismo, proteínas envolvidas nas interações vírus-hospedeiro ou compostos químicos. E todas as redes formadas podem ser visualizadas usando um novo gráfico personalizado “StringStyle“, similar ao padrão usado pelo site STRING.
Para realizar uma busca no stringApp, pode-se utilizar a própria ferramenta do stringApp sendo o “STRING: consulta de proteína” (usa lista de nomes de proteínas, por exemplo, símbolos de genes ou identificadores/números de acesso Uniprot), para obter uma rede STRING das proteínas.
Além disso, há o “STRING: consulta PubMed” (usa uma consulta PubMed com mineração de texto para obter uma rede STRING para as N principais proteínas associadas à consulta). Ademais, tem-se o “STRING: consulta de doença” (usa um nome de doença para recuperar uma rede STRING das principais N proteínas associadas à doença especificada) e o “STITCH: consulta de proteína/composto” (usa uma lista de nomes de proteínas ou compostos para obter uma rede de STITCH).
Para o enriquecimento de vias pelo STRING, o stringApp funciona por meio de uma recuperação do enriquecimento funcional que, ao manter as configurações padrão, sendo necessária apenas a seleção do organismo analisado, como por exemplo para Homo sapiens.
Após a realização da etapa anterior, supracitada, surgirá uma nova aba no painel da tabela sobre o enriquecimento de STRING, onde estará presente os enriquecidos e informações correspondentes para cada categoria de enriquecimento.
Parte 4 – Sugestões de literatura complementar para aprimorar o estudo em biologia de sistemas e redes biológicas.
Visando demonstrar a sugestão de literatura complementar para desenvolver uma reprodutibilidade necessária à Bioinformática e também um contato amplo a conteúdos interessantes promovidos por outros autores, seguem os materiais sugeridos:
- JOYNER, Michael J.; PEDERSEN, Bente K. Ten questions about systems biology. The Journal of Physiology, v. 589, n. 5, p. 1017-1030, 2011.
- MUETZE, Tanja; LYNN, David J. Using the contextual hub analysis tool (CHAT) in cytoscape to identify contextually relevant network hubs. Current protocols in bioinformatics, v. 59, n. 1, p. 8.24. 1-8.24. 13, 2017.
- PILLICH, Rudolf T. et al. NDEx: Accessing Network Models and Streamlining Network Biology Workflows. Current Protocols, v. 1, n. 9, p. e258, 2021.
- PUIG, Rafael Riudavets et al. Network Building with the Cytoscape BioGateway App Explained in Five Use Cases. Current Protocols in Bioinformatics, v. 72, n. 1, p. e106, 2020.
- SAITO, Rintaro et al. A travel guide to Cytoscape plugins. Nature methods, v. 9, n. 11, p. 1069-1076, 2012.
- SALAMON, John; GOENAWAN, Ivan H.; LYNN, David J. Analysis and Visualization of Dynamic Networks Using the DyNet App for Cytoscape. Current protocols in bioinformatics, v. 63, n. 1, p. e55, 2018.
- SU, Gang et al. Biological network exploration with Cytoscape 3. Current protocols in bioinformatics, v. 47, n. 1, p. 8.13. 1-8.13. 24, 2014.
- YEUNG, Natalie et al. Exploring biological networks with Cytoscape software. Current Protocols in Bioinformatics, v. 23, n. 1, p. 8.13. 1-8.13. 20, 2008.
Referências complementares
- CHUANG, Han-Yu; HOFREE, Matan; IDEKER, Trey. A decade of systems biology. Annual review of cell and developmental biology, v. 26, p. 721-744, 2010.
- KITANO, Hiroaki. Computational systems biology. Nature, v. 420, n. 6912, p. 206-210, 2002a.
- KITANO, Hiroaki. Systems biology: a brief overview. Science, v. 295, n. 5560, p. 1662-1664, 2002b.
- KAR, Gozde; GURSOY, Attila; KESKIN, Ozlem. Human cancer protein-protein interaction network: a structural perspective. PLoS computational biology, v. 5, n. 12, p. e1000601, 2009.
- STELZL, Ulrich et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell, v. 122, n. 6, p. 957-968, 2005.
- TOMKINS, James E.; MANZONI, Claudia. Advances in protein-protein interaction network analysis for Parkinson’s disease. Neurobiology of Disease, p. 105395, 2021.
- BARABASI, Albert-Laszlo; OLTVAI, Zoltan N. Network biology: understanding the cell’s functional organization. Nature reviews genetics, v. 5, n. 2, p. 101-113, 2004.
- PAWSON, Tony; LINDING, Rune. Network medicine. FEBS letters, v. 582, n. 8, p. 1266-1270, 2008.
- WINNENBURG, Rainer et al. Facts from text: can text mining help to scale-up high-quality manual curation of gene products with ontologies?. Briefings in bioinformatics, v. 9, n. 6, p. 466-478, 2008.
- WIEGERS, Thomas C. et al. Text mining and manual curation of chemical-gene-disease networks for the comparative toxicogenomics database (CTD). BMC bioinformatics, v. 10, n. 1, p. 1-12, 2009.
- COHEN, K. Bretonnel; HUNTER, Lawrence. Getting started in text mining. PLoS computational biology, v. 4, n. 1, p. e20, 2008.
- WANG, Lucy Lu; LO, Kyle. Text mining approaches for dealing with the rapidly expanding literature on COVID-19. Briefings in Bioinformatics, v. 22, n. 2, p. 781-799, 2021.
- KUMAR, Sunil; KAR, Arpan Kumar; ILAVARASAN, P. Vigneswara. Applications of text mining in services management: A systematic literature review. International Journal of Information Management Data Insights, v. 1, n. 1, p. 100008, 2021.
Conclusão
Neste trabalho foram fornecidos conhecimentos acerca de Biologia de Sistemas, da mineração de texto, da curadoria manual, das redes biológicas, e um tutorial para o desenvolvimento da construção das redes de interação proteína-proteína (redes PPIs) para o desenvolvimento das análises in silico. A partir deste tutorial é válido destacar o quão impactante é o estudo das redes de interação proteína-proteína sobre uma melhor compreensão do aspecto funcional e físico das interações entre as proteínas, assim como, a possibilidade de entender de uma forma mais aprofundada o sistema biológico pesquisado (como um todo) visando à aplicação desses conhecimentos no desenvolvimento de novas terapias e na Medicina Personalizada. Assim, é crucial ter um desenvolvimento desse campo de estudo desde os períodos iniciais das disciplinas biomédicas, biológicas e em áreas que se inter-relacionam com as áreas biológicas e da saúde. Além disso, tornar-se-á importante associar esse campo de estudo também para o ensino básico, objetivando uma maior divulgação e popularização de conteúdos mais aprofundados em uma linguagem mais acessível para a sociedade e a base da pirâmide educacional do país.
Referências
KITANO, Hiroaki. Computational systems biology. Nature, v. 420, n. 6912, p. 206-210, 2002a. ↑
-
CHUANG, Han-Yu; HOFREE, Matan; IDEKER, Trey. A decade of systems biology. Annual review of cell and developmental biology, v. 26, p. 721-744, 2010. ↑
-
KITANO, Hiroaki. Systems biology: a brief overview. Science, v. 295, n. 5560, p. 1662-1664, 2002b. ↑
-
BARABASI, Albert-Laszlo; OLTVAI, Zoltan N. Network biology: understanding the cell’s functional organization. Nature reviews genetics, v. 5, n. 2, p. 101-113, 2004. ↑
-
PAWSON, Tony; LINDING, Rune. Network medicine. FEBS letters, v. 582, n. 8, p. 1266-1270, 2008. ↑
-
KAR, Gozde; GURSOY, Attila; KESKIN, Ozlem. Human câncer protein-protein interaction network: a structural perspective. PLoS computational biology, v. 5, n. 12, p. e1000601, 2009. ↑
-
STELZL, Ulrich et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell, v. 122, n. 6, p. 957-968, 2005. ↑
-
TOMKINS, James E.; MANZONI, Claudia. Advances in protein-protein interaction network analysis for Parkinson’s disease. Neurobiology of Disease, p. 105395, 2021. ↑
-
WINNENBURG, Rainer et al. Facts from text: can text mining help to scale-up high-quality manual curation of gene products with ontologies?. Briefings in bioinformatics, v. 9, n. 6, p. 466-478, 2008. ↑
-
WIEGERS, Thomas C. et al. Text mining and manual curation of chemical-gene-disease networks for the comparative toxicogenomics database (CTD). BMC bioinformatics, v. 10, n. 1, p. 1-12, 2009. ↑
-
KUMAR, Sunil; KAR, ArpanKumar; ILAVARASAN, P. Vigneswara. Applications of text mining in services management: A systematic literature review. International Journal of Information Management Data Insights, v. 1, n. 1, p. 100008, 2021. ↑
-
WANG, Lucy Lu; LO, Kyle. Text mining approaches for dealing with the rapidly expanding literature on COVID-19. Briefings in Bioinformatics, v. 22, n. 2, p. 781-799, 2021. ↑
-
COHEN, K. Bretonnel; HUNTER, Lawrence. Getting started in text mining. PLoS computational biology, v. 4, n. 1, p. e20, 2008. ↑
Acabei de receber minhas análises do espectrômetro de massas já identificadas em 7 bancos de dados diferentes do Uniprot. Se esse tutorial me ajudar (pois, achei ele completíssimo) minha dissertação terá menção honrosa a você meu “xará”!