

Revisão:
BIOINFO – Revista Brasileira de Bioinformática. Edição #. .
DOI:
Docagem molecular é um dos métodos mais populares da modelagem molecular. Essa técnica computacional visa buscar “o encaixe perfeito” entre duas moléculas simulando assim o processo de reconhecimento molecular. A partir da predição de orientação, forma-se um complexo que pode ser usado para estimar a afinidade de ligação ou a força de associação entre as duas moléculas, e com isso, priorizar moléculas que “melhor” se ligam a um parceiro.
O processo de descoberta de novos fármacos é longo, desafiador, cansativo e caro. Esse processo abrange cientistas de diferentes áreas, várias etapas, e um prazo que pode variar de 3 a 20 anos até o medicamento chegar ao mercado. Em 2020, o custo médio para o desenvolvimento de um novo medicamento foi estimado em até US$ 1,3 bilhão [1]. A razão para o alto custo pode ser dividida em três: (i) descoberta da ligação entre alvo molecular e doença confirmando sua importância para a medicina, (ii) ensaios clínicos, e (iii) baixa taxa de sucesso. Em média, apenas 14% de todos os fármacos que começam ensaios pré-clínicos chegam ao mercado [2]. Com isso, os custos com medicamentos que “falharam” são recuperados com os lucros de fármacos aprovados ou inovadores.
A etapa inicial em uma campanha de descobrimento de novos fármacos se caracteriza pela identificação de um alvo molecular, ou vários, que exerçam um papel fundamental no progresso de uma doença. A próxima etapa é identificar moléculas candidatas que parem, inibam, ou revertam o progresso da doença [3]. Para isso são feitas buscas por moléculas que mostrem eficiência biológica em triagens experimentais (in vitro), de larga ou pequena escala, entre múltiplos candidatos. Essas moléculas são chamadas de “hits”. Os “hits” são consequentemente modificados quimicamente, visando melhorar suas propriedades farmacêuticas, como toxicidade, biodisponibilidade e custo-benefício [3]. Essas moléculas são então chamadas de “leads”. Embora os métodos de triagem in vitro permitam a expansão de amostragem do espaço químico, eles costumam apresentar um alto número de falsos positivos e exigir muitos recursos materiais [4]. Além disso, através de ensaios bioquímicos não é possível identificar detalhes atômicos das interações entre alvo e ligante, sendo necessários métodos de resolução estrutural como a cristalografia de raios X, a ressonância magnética nuclear (RMN) e a crio-microscopia eletrônica (Cryo-EM). Esses métodos também são demorados e financeiramente caros por necessitar material, instrumentação específica e pessoas altamente treinadas. Portanto, para auxiliar a triagem biológica, abordagens computacionais são empregadas para priorizar “hits” baseados na conectividade, informações de ligantes ativos, complementaridade geométrica, e predição de afinidade.
Entre os métodos computacionais, a docagem molecular (também conhecida como atracamento, ancoragem, acoplamento ou docking molecular) é a abordagem focada em simular o encaixe entre duas moléculas. Em sua forma mais primitiva, a metodologia baseia-se no conceito de “chave-fechadura” de Emil Fischer proposta em 1894, onde a “chave” (substrato) se encaixa adequadamente no buraco (sítio ativo ou cavidade de ligação) da “fechadura” (enzima ou receptor) para que a reação bioquímica produtiva ocorra [5]. Portanto, chaves muito pequenas, muito grandes ou com entalhes e ranhuras posicionadas incorretamente, não cabem na fechadura. Porém, ao longo dos anos atualizações do conceito de “chave-fechadura”, com por exemplo o conceito do “encaixe induzido”, que pregam flexibilidade conformacional, também foram inseridos na metodologia.

Com isso, pode-se dizer que o conceito da docagem molecular é predizer a orientação preferencial e as interações formadas entre uma molécula em relação a outra, descrevendo de forma computacional o possível complexo alvo-ligante que acontece no ambiente biológico. A metodologia de docagem foi introduzida em 1982 por Kuntz e colaboradores [6] no artigo intitulado “A Geometric Approach to Macromolecule-Ligand Interactions”. Kuntz e colaboradores descrevem a ideia de que o reconhecimento molecular entre moléculas, químico e geométrico, pode ser explorado através de modelos tridimensionais tanto do ligante e quanto do alvo. Desde então, a docagem molecular é empregada como uma maneira rápida de estimar o modo de ligação de um determinado composto dentro de um alvo e para prever a afinidade dessa ligação [7].
Tradicionalmente, os chamados alvos são macromoléculas (proteína, DNA/RNA, peptídeos), enquanto os ligantes são pequenas moléculas (fármacos, ligantes endógenos). Hoje em dia, métodos de docagem entre macromoléculas também são comuns. Os experimentos computacionais de docagem se iniciam após a obtenção das estruturas tridimensionais de alvo e ligante. Estruturas dos alvos são obtidas de bancos de dados de estruturas tridimensionais como o PDB [8] ou podem ser modeladas por modelagem comparativa. Já as estruturas dos ligantes podem ser geradas através de programas especializados ou de bancos de dados de ligantes como o ZINC [9] e Pubchem [10]. Obtidos esses elementos, vários programas de docagem disponíveis podem ser utilizados para ajustar o ligante em uma região definida do alvo, normalmente, um sítio ativo ou uma cavidade de interesse. Esses programas combinam e otimizam variáveis como complementaridade estérica, hidrofóbica e eletrostática, e estimam a afinidade de ligação através de uma função de pontuação [11].
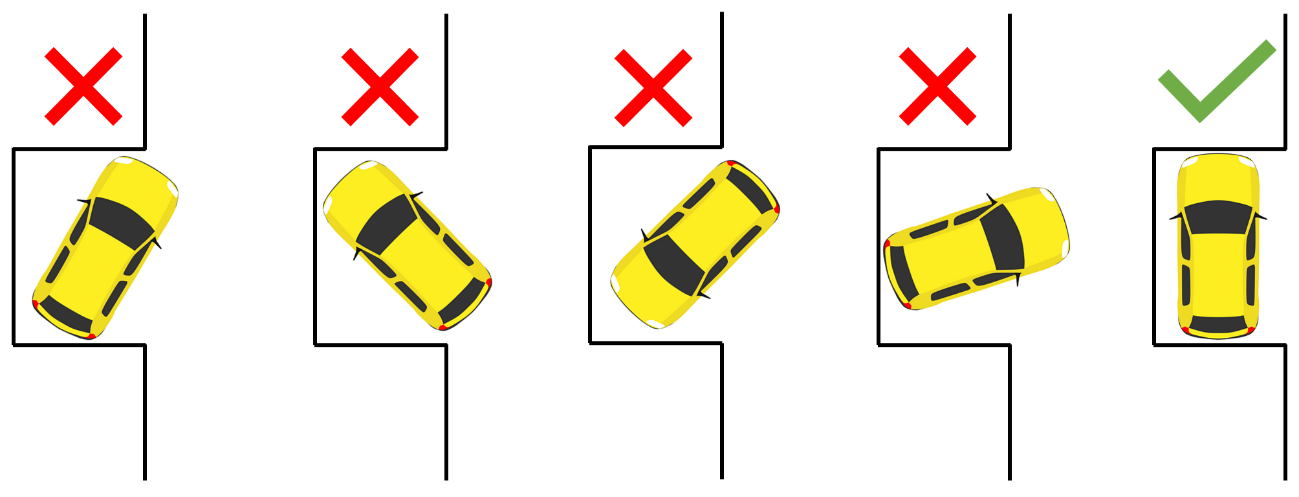
Todos os programas possuem dois componentes essenciais: um bom algoritmo de posicionamento e um sistema robusto de classificação ou pontuação. Um protocolo de docagem requer extensa amostragem de espaço conformacional do ligante para posicioná-lo no sítio de ligação de uma proteína e, portanto, um grande número de potenciais orientações e conformações de um ligante são geradas, as chamadas poses. Um bom algoritmo de posicionamento amostra “todos” os modos de ligação possíveis, enquanto o sistema de pontuação classifica todas as soluções e identifica o “modo de ligação” mais provável do ligante [5]. Podemos pensar nesse processo como um aluno de uma autoescola aprendendo a fazer baliza, onde várias tentativas são feitas até o carro ficar perfeitamente alinhado na vaga (Figura 2).
Apesar de soar simples, os processos de geração e ranqueamento de poses são problemas complexos e desafiadores. Posicionar o ligante dentro do espaço delimitado do sítio de ligação é um processo exaustivo de busca de orientações e mapeamento das interações entre os resíduos e o ligante [5]. Normalmente, esse processo requer um equilíbrio entre tempo e acurácia, ou seja, o maior número de soluções deve ser explorado mantendo um tempo computacional eficiente, já que em um processo de triagem centenas a milhões de ligantes são utilizados. Enquanto isso, a função de pontuação deve ser suficientemente eficiente de forma a predizer a afinidade de ligação entre alvo e ligante ou pelo menos conseguir pontuar o modo de ligação mais próximo ao experimental possível [12]. Para manter esse equilíbrio, simplificações são impostas, tanto nos algoritmos de busca para gerar os modos de ligação das moléculas, quanto nas funções de pontuação pelos programas de docagem.
Por exemplo, muitos programas não consideram moléculas de água ou a presença de solvente na formação do complexo. A ausência de solvente é compreensível por inserir muitas moléculas extremamente flexíveis, onde a mudança de orientação de uma molécula apenas afeta as moléculas vizinhas e a rede de interações entre elas [7]. Portanto, moléculas de águas, normalmente, não são explicitamente consideradas, apesar de algumas funções de pontuação avaliarem efeitos do solvente em seus termos. Outra limitação do método é a falta de movimento, onde tipicamente apenas o ligante é considerado flexível e o alvo rígido. Na vida real, o evento de ligação entre alvo e ligante não é estático como na docagem, e sim dinâmico, com ambos sofrendo múltiplos rearranjos. Recentemente, abordagens que consideram o alvo flexível têm sido disponibilizadas, como docagem em múltiplas conformações do alvo ou considerando como rotacionáveis as cadeias laterais de resíduos selecionados, mas sacrificando a rapidez do método (e não necessariamente ganhando robustez) [13]. O problema mais difundido na metodologia talvez seja a simplificação das interações (incluindo interações iônicas, van der Waals e de hidrogênio) entre alvo e ligante, levando a estimativa de afinidade entre esses elementos a não ser confiável. Os termos em uma função de pontuação são simples funções de energia potencial, geralmente relacionadas a campos de força ou potenciais estatísticos [7]. Com isso, efeitos de polarização ou a presença de prótons não são considerados de forma a agilizar a abordagem.
Apesar de suas simplificações, a docagem molecular é uma das abordagens mais populares dentro do processo de descoberta de novos fármacos com auxílio de métodos computacionais. Portanto, mesmo com os desafios a serem superados, a metodologia é amplamente utilizada na priorização de compostos “hit” em uma triagem (virtual) de múltiplos compostos ou para otimização de compostos “lead”. Podendo então explorar possibilidades e hipóteses de forma rápida e com custo baixo quando comparados com experimentos de bancada. Além disso, cada vez mais a metodologia tem sido usada em conjunto com outros métodos, como dinâmica molecular [14], aprendizado de máquina [15], mecânica quântica [16], entre outros. Tudo isso tem permitido validar e melhorar a qualidade dos resultados, ao invés de contar apenas com a docagem. Sem dúvida, as técnicas computacionais são essenciais para os avanços científicos na compreensão da formação de complexos. Porém, muito cuidado deve ser tomado ao assumir resultados computacionais como “perfeitos”, em grande parte pelas várias simplificações. Além disso, sempre que possível deve-se buscar o emprego de técnicas experimentais em conjunto.
Referências
1. Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018 [Published March 3, 2020]. JAMA.
2. Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of Clinical Trial Success Rates and Related Parameters. Biostatistics 2019, 20, 273–286, doi:10.1093/biostatistics/kxx069.
3. Muntha, P. Drug Discovery & Development–A Review. Res. & Rev.: J. Pharm. Pharmaceut. Sci 2016, 5, 135–142.
4. Yan, X.C.; Sanders, J.M.; Gao, Y.-D.; Tudor, M.; Haidle, A.M.; Klein, D.J.; Converso, A.; Lesburg, C.A.; Zang, Y.; Wood, H.B. Augmenting Hit Identification by Virtual Screening Techniques in Small Molecule Drug Discovery. Journal of chemical information and modeling 2020, 60, 4144–4152.
5. Tripathi, A.; Bankaitis, V.A. Molecular Docking: From Lock and Key to Combination Lock. Journal of molecular medicine and clinical applications 2017, 2.
6. Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A Geometric Approach to Macromolecule-Ligand Interactions. Journal of molecular biology 1982, 161, 269–288.
7. Pantsar, T.; Poso, A. Binding Affinity via Docking: Fact and Fiction. Molecules 2018, 23, 1899.
8. Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Research 2000, 28, 235–242, doi:10.1093/nar/28.1.235.
9. Sterling, T.; Irwin, J.J. ZINC 15 – Ligand Discovery for Everyone. Journal of Chemical Information and Modeling 2015, 55, 2324–2337, doi:10.1021/acs.jcim.5b00559.
10. Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B. PubChem 2019 Update: Improved Access to Chemical Data. Nucleic acids research 2019, 47, D1102–D1109.
11. Sethi, A.; Joshi, K.; Sasikala, K.; Alvala, M. Molecular Docking in Modern Drug Discovery: Principles and Recent Applications. Drug Discovery and Development-New Advances 2019, 1–21.
12. Spyrakis, F.; Cozzini, P.; Kellogg, G.E. Docking and Scoring in Drug Discovery Burger’s Medicinal Chemistry and Drug Discovery. 7th 2009.
13. Chen, Y.-C. Beware of Docking! Trends in pharmacological sciences 2015, 36, 78–95.
14. Santos, L.H.S.; Ferreira, R.S.; Caffarena, E.R. Integrating molecular docking and molecular dynamics simulations. In Docking screens for drug discovery; Springer, 2019; pp. 13–34.
15. Khamis, M.A.; Gomaa, W.; Ahmed, W.F. Machine Learning in Computational Docking. Artificial intelligence in medicine 2015, 63, 135–152.
16. Adeniyi, A.A.; Soliman, M.E.S. Implementing QM in Docking Calculations: Is It a Waste of Computational Time? Drug discovery today 2017, 22, 1216–1223.
[…] Vencedor(a): Lucianna Santos – “Docagem molecular: em busca do encaixe perfeito e acessível” […]