

Desenvolvido pela DeepMind em 2020, o AlphaFold é uma inovadora ferramenta de inteligência artificial que surgiu na CASP14, uma competição de predição de estruturas proteicas. À época, ele foi apresentado como uma solução para o desafiante problema do enovelamento de proteínas. Esse problema envolve a compreensão de como uma sequência de aminoácidos se converte em uma estrutura tridimensional. Uma proteína não enovelada vai mudando de conformação, diminuindo a entropia até chegar no estado de menor energia, em que ela estará em seu estado nativo. O “paradoxo de Levinthal” destaca a complexidade desse processo, sugerindo que, embora uma proteína possa se dobrar em milissegundos, o tempo necessário para calcular todas as estruturas possíveis é maior do que a idade do universo conhecido. Embora o AlphaFold não tenha resolvido completamente esse desafio, ele marcou um avanço significativo ao prever com precisão as estruturas proteicas a partir de sequências primárias, revolucionando a pesquisa em biologia.
Neste artigo, nossa intenção será elucidar o processo pelo qual esse sistema constrói as estruturas tridimensionais, abordando também aulas práticas sobre modelagem molecular, utilizando o AlphaFold como ferramenta central em nossos experimentos. Com isso, esperamos proporcionar uma compreensão mais aprofundada das capacidades dessa tecnologia e seu potencial impacto no avanço da pesquisa em bioinformática e biologia molecular.
Introdução
AlphaFold é uma ferramenta altamente sofisticada que prevê estruturas de proteínas através de outras já conhecidas, baseada em redes neurais profundas. Até então, já realizou mais de 200 milhões de predições [1], tendo sido treinada com estruturas experimentais das proteínas disponíveis no Protein Data Bank (PDB) [2]. Sua primeira versão, AlphaFold 1, foi construída em 2018, com base no trabalho desenvolvido por várias equipes anteriores. Elas tentavam encontrar mudanças em diferentes resíduos que pareciam estar correlacionados, embora não fossem consecutivos na cadeia principal. Tais correlações sugeriam que os resíduos poderiam estar próximos fisicamente, embora não próximos na sequência, e isso permitiu que os cientistas estimassem um mapa de contatos baseado nessas informações [3, 4]. O AlphaFold 1 estendeu isso para estimar uma distribuição de probabilidade de quão próximos os resíduos poderiam estar, construindo um mapa de distâncias prováveis. Assim, o AlphaFold 1 é um preditor de mapas de distância implementado como redes neurais profundas. Juntamente com um mapa de distância na forma de um histograma, o AlphaFold prevê ângulos Φ e Ψ (Figura 1) para cada resíduo, que são usados para criar a estrutura 3D inicial prevista.
Os ângulos descritos acima são de torção em torno das ligações peptídicas, de forma que o ângulo Phi (Φ) é medido entre o átomo de nitrogênio (N) e o átomo de carbono α (Cα), enquanto o ângulo Psi (Ψ) é medido entre o átomo de carbono α (Cα) e o átomo de carbono do grupo carbonila (C=O). Ambos possuem um papel importante na conformação proteica, uma vez que esses ângulos são restritos devido a limitações estéricas e interações eletrônicas. Essas variações nos ângulos contribuem para a diversidade estrutural de proteínas, levando a diferentes configurações tridimensionais e, consequentemente, influenciando suas propriedades funcionais.
O gráfico de Ramachandran, gráfico comumente utilizado para verificar a qualidade das estruturas, representa o espaço conformacional das proteínas, definindo as regiões permitidas e proibidas com base nas conformações estericamente aceitáveis das ligações peptídicas [5]. Dessa forma, a compreensão dos ângulos Phi e Psi é fundamental na predição da estrutura proteica e alterações conformacionais, permitindo, inclusive, a modificação racional de proteínas a fim de melhorar sua estabilidade, atividade catalítica e afinidade por ligantes [6].

Figura 1. Ilustração dos ângulos Phi (Φ) e Psi (Ψ). Fonte: Fang, C. et al. (2018) [7], disponível em: 10.1109/TCBB.2018.2814586. Acesso em 31/07/2023.
AlphaFold 2
Em 2021, foi criada a segunda versão do AlphaFold, uma vez que a equipe do DeepMind havia identificado que sua abordagem anterior tendia a superestimar as interações entre os resíduos que estavam próximos na sequência em comparação com as interações entre os resíduos mais distantes ao longo da cadeia. Como resultado, o AlphaFold 1 poderia preferir modelos com uma estrutura um pouco mais secundária (alfa-hélices e folhas-beta) do que na realidade [8, 9, 10]. Assim, o AlphaFold 2 surgiu como uma inovação do primeiro, baseado no reconhecimento de padrões de estruturas e sequências. Vale ressaltar que toda a informação sobre o funcionamento da ferramenta pode ser acessada no artigo “Highly accurate protein structure prediction with AlphaFold” [11], referente à sua segunda versão
Para utilizar a ferramenta, deve-se realizar a instalação local usando o código open-source disponível no site da DeepMind através do link https://www.deepmind.com/open-source/alphafold, sendo necessário verificar o tipo de sistema e quantidade de memória necessários para uso (requer placa de vídeo).
Entretanto, pode-se ainda utilizar uma versão online disponível através do Google Colab, denominada ColabFold [12], através do seguinte link: https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb. Nela, utiliza-se apenas a sequência como input e pode-se alterar alguns parâmetros que julgar necessários.
Como funciona essa ferramenta?
A metodologia da ferramenta pode ser vista na Figura 2, e consiste de três partes:
- 1. Pré-processamento
- 2. Evoformer
- 3. Construção da estrutura
A figura abaixo resume todo o processo:

Figura 2. Esquema representando a metodologia utilizada pelo AlphaFold 2. Fonte: adaptado de: Jumper, J. et al. (2021) [12], disponível em: https://doi.org/10.1038/s41586-021-03819-2. Acesso em 31/07/2023.
Pré-processamento
É a primeira etapa do AlphaFold e tem como objetivo preparar a sequência de aminoácidos da proteína para a predição da sua estrutura tridimensional. Seu esquema de funcionamento pode ser visto com maior clareza na Figura 3, abaixo.

Figura 3. Esquema representando a fase de pré-processamento da metodologia utilizada pelo AlphaFold 2. Adaptado de: Jumper, J. et al. (2021) [12], disponível em: https://doi.org/10.1038/s41586-021-03819-2. Acesso em 31/07/2023.
Essa etapa consiste em:
-
A ferramenta recebe como entrada a sequência da proteína, que é então dividida em segmentos estruturados e não estruturados. Os segmentos estruturados são aqueles que possuem uma estrutura 3D bem definida e estável, como alfa-hélices, folhas-beta e regiões de loop, ao passo que os não estruturados são mais flexíveis. Dessa forma, os segmentos não estruturados são removidos da sequência.
-
A sequência é utilizada para realizar uma busca em bancos de dados genéticos e de estruturas, a fim de encontrar sequências semelhantes para que possa ter uma base para a criação da estrutura. Em seguida, as sequências semelhantes são alinhadas com a sequência de interesse para gerar um alinhamento múltiplo de sequências (MSA), que permite ver a similaridade entre elas e determinar quais são mais semelhantes. O MSA é filtrado para remover sequências redundantes e de baixa qualidade, garantindo apenas sequências mais confiáveis na predição da estrutura.
-
O resultado final da primeira etapa é uma representação do MSA, uma matriz Nseq x Nres, onde Nseq é o número de sequências na MSA e Nres é o número de resíduos na sequência de aminoácidos; além de uma representação de pares: uma matriz Nres x Nres, onde cada elemento representa a relação entre dois resíduos, mostrando os prováveis aminoácidos que estarão em contato com os outros.
Uma observação importante é que muitas proteínas desempenham funções similares em diversas espécies por compartilharem um ancestral comum. Apesar das sequências sofrerem mutações ao longo do tempo, a sua estrutura tende a permanecer semelhante, uma vez que mudanças bruscas podem desestabilizar uma estrutura e possivelmente inviabilizar a função desempenhada. Essa informação ganha relevância no contexto do AlphaFold, já que a ferramenta utiliza sequências e estruturas de proteínas de espécies semelhantes. Essa escolha é estratégica, pois o AlphaFold busca por alinhamentos similares em bancos de dados genéticos para criar sua representação do MSA, de forma a projetar uma estrutura baseada nas sequências similares à sequência de entrada. Trazendo um exemplo atual, a proteína Spike do SARS-CoV-2, tão falada nos últimos anos, é muito semelhante à proteína Spike do vírus SARS-CoV, responsável por uma epidemia em 2002. Há pouca diferença entre os resíduos de aminoácidos entre as duas proteínas, e suas estruturas são muito semelhantes. Provavelmente antes de termos a estrutura experimental da proteína do SARS-CoV-2, o AlphaFold utilizaria a proteína do SARS-CoV como base para gerar sua estrutura teórica.
Evoformer
A segunda etapa é composta pelo processamento das entradas através de camadas repetidas de um bloco de redes neurais chamado Evoformer, um transformador, e pode ser vista com maior clareza na Figura 4, abaixo.

Figura 4. Esquema representando a fase do Evoformer da metodologia utilizada pelo AlphaFold 2. Fonte: adaptado de: Jumper, J. et al. (2021) [12], disponível em: https://doi.org/10.1038/s41586-021-03819-2. Acesso em 31/07/2023.
As representações do MSA e de pares de resíduos, geradas na primeira etapa, são utilizadas como entrada no Evoformer. A representação do MSA gerada pelo Evoformer é uma versão refinada do MSA original, que leva em consideração as informações evolutivas e a relação entre as sequências no alinhamento, ou seja, ele filtra as informações mais relevantes das representações geradas na etapa anterior. A matriz gerada nesta etapa é utilizada para gerar uma representação tridimensional da proteína, levando em consideração a relação espacial entre os resíduos. Resumindo, como saída, ele tem representações melhoradas daquelas que foram utilizadas como entrada, que serão usadas na próxima etapa. Cada rede do AlphaFold possui 48 blocos do Evoformer e, dependendo da estrutura, pode passar diversas vezes até chegar em um resultado satisfatório, processo denominado reciclagem.
Módulo de estrutura
É a terceira etapa e tem como objetivo gerar a representação tridimensional da proteína a partir das informações processadas pelo Evoformer. É possível ver seu funcionamento com clareza na Figura 5.

Figura 5. Esquema representando a fase do módulo de estrutura da metodologia utilizada pelo AlphaFold 2. Fonte: adaptado de: Jumper, J. et al. (2021) [2], disponível em: https://doi.org/10.1038/s41586-021-03819-2. Acesso em 31/07/2023.
O mapa de representações gerado na etapa anterior descreve as probabilidades de distância entre os pares de átomos presentes na proteína, ou seja, um mapa de distâncias. O módulo estrutural do AlphaFold utiliza um sistema de redes neurais, mais precisamente, oito blocos neurais, que são aplicados em série para traduzir o perfil de probabilidade de distância em uma estrutura tridimensional. Esta rede neural é treinada com um grande conjunto de dados de proteínas com estruturas tridimensionais conhecidas. Durante o treinamento, a rede aprende a mapear os perfis de probabilidade de distância para estruturas tridimensionais que são consistentes com esses perfis. Cada bloco neural recebe como entrada a representação 3D gerada pelo bloco anterior e gera uma nova, dessa vez refinada. O resultado final é uma estrutura tridimensional prevista para a proteína, com cores que representam a confiabilidade de cada região da estrutura. Além disso, essa etapa também pode passar pelo processo de reciclagem, melhorando cada vez mais a estrutura.
AlphaFold 2 x ColabFold
Até o momento, entendemos que o AlphaFold 2 trata-se de um software baseado em IA para realizar a predição da estrutura 3D de uma proteína. Existem três práticas experimentais confiáveis para saber como é a conformação da proteína: cristalografia e difração de raios X, ressonância magnética nuclear (NMR) e microscopia eletrônica criogênica. No entanto, essas técnicas são trabalhosas, custosas e demoradas, sem contar a necessidade de mão de obra especializada. Partindo deste princípio, os cientistas vêm trabalhando há tempos para realizar previsões sobre a estrutura 3D a partir de diversos métodos computacionais. Eles obtiveram sucesso apenas quando a DeepMind lançou o AlphaFold, alcançando até 90% de precisão [13]! Isso é fantástico!
Como disponibilizado anteriormente, o AlphaFold 2 possui seu código disponível no Github e é possível compilar no próprio computador, porém, é necessário utilizar equipamentos robustos devido ao custo computacional e operacional da IA. A título de conhecimento, é necessário que o computador tenha pelo menos 3 TB de armazenamento para baixar o banco de dados do AlphaFold, além de placas de vídeo da NVIDIA. Com isso, a DeepMind e o Instituto Europeu de Bioinformática (EMBL-EBI) se uniram para resolver este problema! Essa parceria resultou em um banco de dados denominado AlphaFold DB (https://alphafold.ebi.ac.uk/), que disponibiliza gratuitamente 200 milhões de previsões de estruturas do proteoma humano e de outros 47 organismos importantes na pesquisa da saúde global. É possível baixar este repositório acessando o UNIPROT (repositório padrão de sequências e anotações de proteínas). Assim, basta pesquisar pelo código UNIPROT da proteína de interesse e baixar diretamente pela plataforma. Entretanto, uma dificuldade comum na rotina de bioinformatas é de, muitas vezes, não ter disponíveis informações sobre o nome da proteína/gene, ou até mesmo o código UNIPROT, mas apenas um trecho de uma sequência. Neste contexto, é possível procurar esta sequência em um banco de dados, em busca de sequências semelhantes.
Mas, se quisermos predizer e visualizar a estrutura e não tivermos como rodar o Alphafold em um sistema computacional comum, como proceder? Se você pensou em ColabFold, acertou! Com ColabFold é possível predizer estruturas de maneira simplificada utilizando o notebook Colab, com pouca diferença da precisão do AlphaFold 2. A depender do tamanho da proteína, em poucos instantes a estrutura será prevista pelo próprio ColabFold e estará pronta para ser baixada em seu sistema.
Agora que você já tem uma noção das diferenças, pode aprender com a gente com duas práticas de modelagem, a seguir.
Prática de modelagem
Neste primeiro tópico prático, começaremos a utilizar esta ferramenta poderosíssima para aprimorar nossa compreensão sobre a modelagem de proteínas. Porém, antes de começarmos nossa primeira prática, é necessário possuir conceitos biológicos básicos sobre o processo de enovelamento de proteínas e as condições físico-químicas para a sua configuração final.
Etapa 1: Prepare seus dados
Para obter a sequência primária, acessaremos o NCBI (National Center for Biotechnology Information) através do link https://www.ncbi.nlm.nih.gov/. Selecionamos o campo “Protein” (passo 1) e iremos escrever no campo de busca o nome da proteína de interesse, neste caso, vamos trabalhar com a “superoxide dismutase” (passo 2). Em seguida, clicamos no botão “Search” (passo 3). Já na página do resultado da nossa busca, selecionamos o organismo de interesse (“Plants” – passo 4). Neste caso, vamos trabalhar com a proteína superóxido dismutase na soja (Glycine max – passo 5). Esta enzima tem o papel importante na resposta ao estresse oxidativo nas plantas diante da ação de um herbicida, por exemplo.

Figura 6. Página do NCBI mostrando o resultado na nossa primeira busca para a proteína de interesse.
Utilizaremos o primeiro resultado da nossa busca e, ao clicar no nome da proteína (passo 5), abrirá uma segunda página. Nessa página, você pode encontrar informações importantes para esta proteína. Como nosso objetivo é obter a sequência primária, vamos clicar no botão “FASTA” (passo 6).
Pronto! Agora, é só copiar e colar a sequência em um bloco de notas.
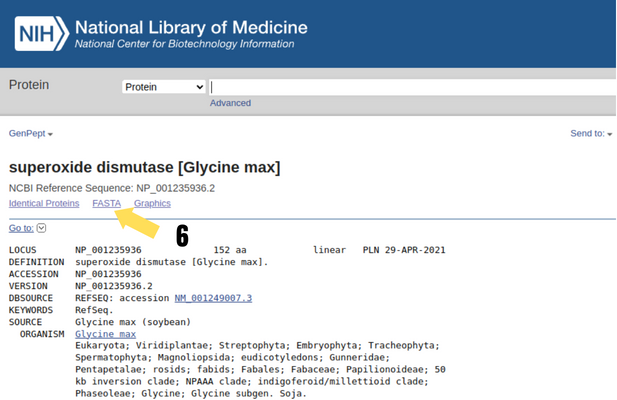
Figura 7: Nesta seção, você consegue encontrar informações gerais da proteína de interesse, por exemplo: seu locus gênico, tamanho, autores da descoberta, comentários gerais, etc.
Etapa 2: Acesse e execute o AlphaFold 2
Para esta etapa, é necessário que você possua uma conta registrada no Google para o uso do AlphaFold 2 no Google Colab.
Para começar, no campo de busca do navegador, acesse o link: https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb e clique em “Conectar” e “ok” para gerar o aviso do uso (passo 7).
Figura 8: O Google Colaboratory ou Colab, é um serviço disponibilizado pela própria Google. Esta ferramenta permite rodar códigos em Python em uma máquina google através da tecnologia Cloud Computing.
Selecionamos a sequência primária obtida na Etapa 1 e colamos no campo de query_sequence. Por fim, selecionamos o botão Control + F9 para executar o código inteiro. Outra opção é executar cada célula de código individualmente, permitindo a visualização de cada etapa separadamente. É importante esperar que cada célula acabe de ser executada antes de iniciar a próxima.
Fique atento pois, para este tutorial, usaremos apenas a sessão de “query_sequence” e “jobname”; os outros parâmetros da plataforma não serão utilizados e, portanto, não precisam ser modificados. No caso, “MSA options” diz respeito a parâmetros do alinhamento múltiplo de sequências e em “Advanced settings” o usuário pode modificar algumas configurações avançadas, como, por exemplo, o número de reciclagens a serem realizadas (explicado anteriormente no funcionamento do AlphaFold) ou o “dpi”, que é basicamente a qualidade da imagem a ser gerada.

Figura 9: Neste campo, há vários parâmetros de seleção. Para esta prática, utilizaremos apenas dois: query_sequence e jobname.
Etapa 3: Analisar e interpretar os resultados
Depois de pedir para executar, a etapa da modelagem pode levar algum tempo, dependendo do tamanho da sua sequência. No nosso caso, pode levar até 5 minutos, uma vez que a proteína possui 152 aminoácidos. Ao finalizar, o próprio sistema pedirá que salve o resultado no computador. No entanto, para nossa breve análise, não precisaremos baixar o resultado, já que ele pode ser facilmente visualizado no próprio Colab no final da página. Não se preocupe com os seguintes tópicos do Colab, como Install dependencies, Run Prediction, Display 3D structure e Plots. Eles se referem aos campos que o próprio Colab utilizará, ou seja, executarão de modo automático.
Pronto! No final da execução teremos nossa proteína de interesse com a sua modelagem predita. Para gerar o modelo tridimensional predito (Figura 11), basta visualizar a próxima etapa “Display 3D structure”, juntamente com o nível de confiança de cada região modelada.

Figura 10: Resultado final da predição da proteína superóxido dismutase da soja Glycine max L.
No campo de “Plots”, podemos visualizar alguns gráficos que indicam a qualidade da estrutura. O primeiro parâmetro é constituído por quatro gráficos e é denominado erro de alinhamento previsto (PAE), que consiste na avaliação da confiança em relação à posição no enovelamento dos domínios proteicos. Esse dado será abordado em nossa segunda prática, a modelagem de complexos, pois não serve para proteínas com apenas um domínio. O gráfico abaixo, na esquerda, mostra o número de sequências por posição, como mostrado na Figura 10 (A). Esse gráfico mostra a cobertura da sequência tendo como base todas as sequências semelhantes encontradas, que são representadas no eixo Y, enquanto o eixo X mostra as posições dos resíduos nas sequências. Se houver um gap entre a sequência de entrada e as sequências encontradas, ou seja, regiões faltantes, o trecho faltante estará representado como uma região em branco, indicando uma baixa cobertura. Ao lado, há uma faixa vertical colorida indicando a identidade da sequência com a de entrada, ou seja, uma indicação de similaridade. Além disso, o AlphaFold produz uma estimativa de confiança por resíduo em uma escala de 0 a 100, que estará presente nos arquivos quando você baixar os resultados. Essa medida de confiança é chamada de Local Distance Difference Test (plDDT), (Figura 10 B), onde a confiabilidade é estimada por resíduo (azul para alta confiança, vermelho para baixa confiança). Espera-se que regiões com plDDT maior que 90 sejam modeladas com alta precisão. Por outro lado, regiões com plDDT entre 50 a 70 indicam baixa confiança e devem ser tratadas com cautela. Abaixo de 50, pode indicar regiões que não podem ser interpretadas [1]. Na legenda, são mostrados os “ranks” de 1 a 5, que são os cinco modelos estruturais da sequência que foram gerados pelo Alphafold. Os autores discutem melhor no artigo da ferramenta.

(A) (B)
Figura 11: Gráficos mostrando o alinhamento múltiplo de sequências por resíduo (A), ou seja, o número médio de leituras que se alinham ou “cobrem” bases da referência (sequência de input), e a estimação confiabilidade por residuo (B).
Arquivos gerados:
Ao final da prática, você também possui a opção de salvar os resultados da sua modelagem. Para isso, basta rodar a célula “Package and download results”, onde você pode salvar o arquivo em formato .zip onde desejar. Ao descompactá-lo, terão diversos arquivos dentro da pasta, dos quais você utilizará para sua análise:
-
O arquivo “nomeDaEstrutura_coverage.png”, contendo o gráfico de cobertura de sequências (sequence coverage);
-
O arquivo “nomeDaEstrutura_pae.png”, contendo os gráficos de erro de alinhamento predito (PAE);
-
O arquivo “nomeDaEstrutura_plddt.png”, contendo o gráfico de Local Distance Difference Test;
-
Os arquivos das estruturas geradas, em formato .pdb. Eles estarão nomeados como “nomeDaEstrutura_unrelaxed_rank_001_alphafold2_ptm_model_5_seed_000.pdb”, sendo numerados de acordo com a classificação de melhor estrutura.
Etapa 4: Validação das estruturas previstas (Bônus)
Após obter o modelo predito da proteína, é necessário verificar suas semelhanças e scores (pontuações) de modelagem no PDB. No nosso caso, o modelo ainda não foi determinado pelos métodos convencionais (Cristalografia e difração de raio-X, Ressonância Magnética Nuclear ou Cryo-EM). Assim, é importante realizar métodos de alinhamento de sequências tridimensionais por proteínas homólogas a esta que já se encontram resolvidas.
Independente do nosso resultado, a etapa final de validação é crucial para o aprofundamento do nosso estudo e novos achados, pois compara o resultado obtido com dados experimentais, localização dos resíduos de sítios ativos, ligação de ligantes e outros detalhes. Ao validar este resultado através de comparações, e utilizando algumas ferramentas comumente utilizadas para este fim, garantimos maior qualidade e confiabilidade à pesquisa. Algumas dessas ferramentas incluem:
-
MolProbity (https://pubmed.ncbi.nlm.nih.gov/29067766/), que identifica problemas de geometria e estereoquímica;
-
Verify3D (https://www.doe-mbi.ucla.edu/verify3d/), que compara a proteínas com outras já bem resolvidas;
-
Prosa (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1933241/), que identifica conformações atípicas na estrutura.
Prática de modelagem de complexos
Iniciando a prática, é importante ressaltar que o AlphaFold não realiza o docking molecular propriamente dito. Diferentemente das ferramentas de docking comumente usadas, não calcula a afinidade de ligação entre as moléculas, como também não busca, no espaço conformacional, a melhor conformação por meio de algoritmos de busca. O que a ferramenta realiza é a modelagem de ambos os elementos, tanto receptor (proteína) quanto do ligante (proteína/peptídeo) em complexo, tentando modelar a região de ligação entre receptor-ligante.
Para exemplificar, realizaremos a modelagem do complexo TNF-α, uma citocina pró-inflamatória de fundamental importância para o processo de defesa do organismo, com um de seus receptores, TNR1 [14, 15].
Etapa 1: Adquirindo as sequências
Para iniciar o tutorial, o primeiro passo é baixar as sequências das proteínas que usaremos. Nesta etapa vamos acessar o PDB (Protein Data Bank), no endereço https://www.rcsb.org/. Depois, vamos buscar pela estrutura de TNF-α, utilizando o identificador 1TNF (Figura 12).

Figura 12. Buscando pela estrutura de TNF-α.
Após encontrarmos a estrutura no banco de dados, vamos baixar a sequência de aminoácidos. Para isso, clique na primeira estrutura encontrada. Depois de abrir, você deve clicar no botão lateral Download Files. Em seguida, clique em Fasta Sequence para baixar o arquivo com a sequência de aminoácidos (Figura 13). Após baixarmos a sequência de TNF-α, vamos realizar o mesmo procedimento para o receptor TNFR1, nesse caso vamos buscar ar pelo ID 1TNR no PDB, e baixar a sequência selecionando a sequência correspondente a cadeia B.

Figura 13. Baixando a sequência de TNF-α.
Etapa 2: Modelando o complexo
Após baixar as sequências, vamos começar a modelar o complexo. Para isso, abriremos o ColabFold. Em seguida, vamos colar as sequências na aba query_sequence. Como a TNF-α é um trímero, vamos colar a sequência da citocina três vezes, separando cada sequência utilizando “:” (dois pontos). A seguir, vamos colar a sequência do receptor 1TNR na aba query_sequence, após a sequência de TNF-α. Após colar a sequência, podemos inserir um nome no trabalho na aba “jobname”. Clique em “Ambiente de execução” e depois clique em “Executar tudo”. Vamos aguardar que todo o script seja executado para analisar os resultados.

Figura 14. Iniciando a modelagem do complexo.
Etapa 3: Avaliando os resultados
O primeiro gráfico a ser observado é o da Figura 15. Nele, podemos observar a cobertura das sequências encontradas como template para modelagem. Algo importante a se observar é que, diferentemente da modelagem de uma proteína de cadeia única, o gráfico é dividido entre as cadeias. No caso do nosso gráfico, ele está dividido em quatro cadeias: as três primeiras correspondem a cadeias da proteína TNF-α e a última cadeia corresponde à TNF-α. Na parte superior, encontramos mais um bloco de cobertura, relacionado às regiões de ligação entre as cadeias das proteínas. Nota-se que o ColabFold encontrou sequências com boa cobertura, o que nos leva a crer que a modelagem será bem executada.
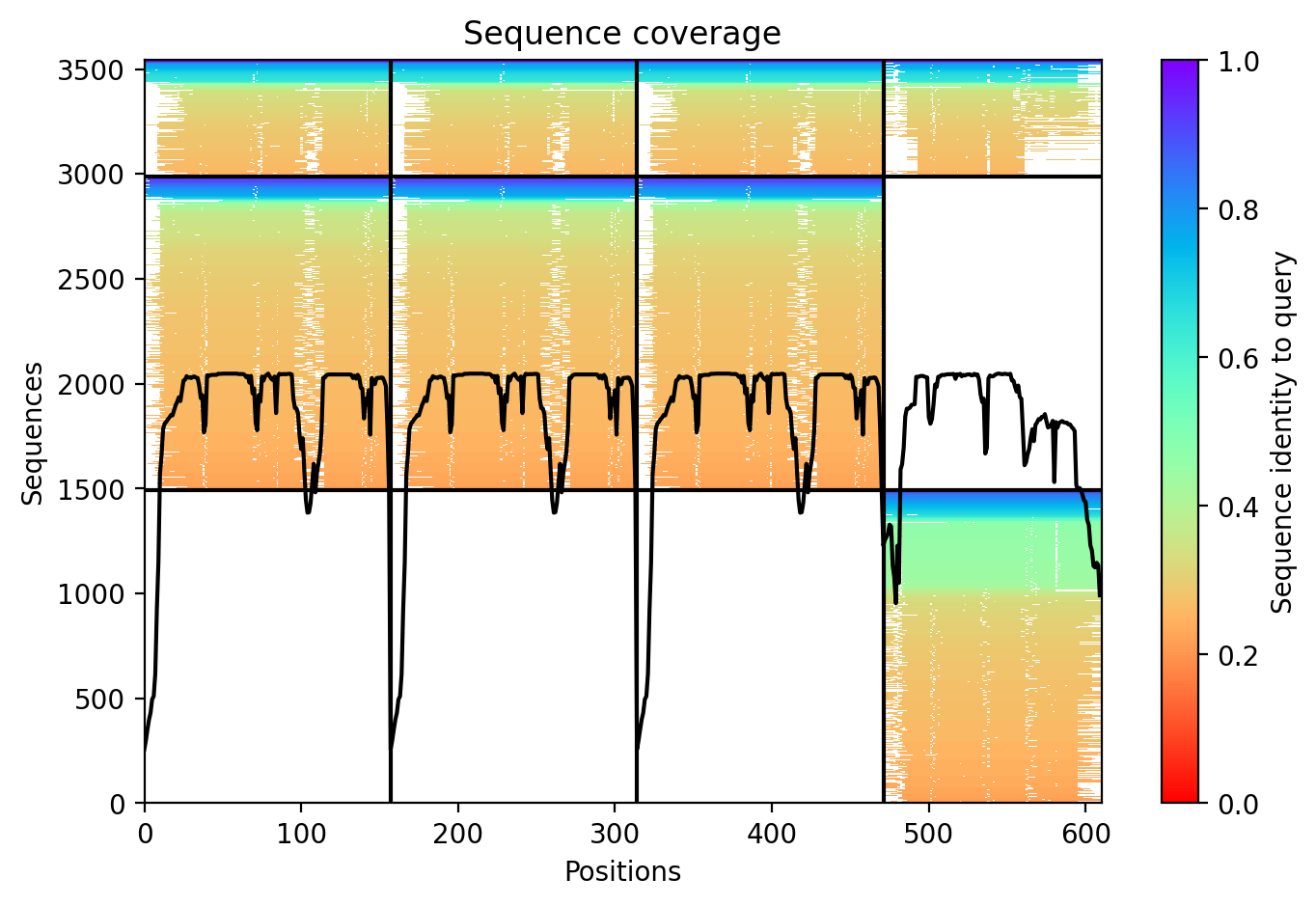
Figura 15. Gráfico de cobertura das sequências de entrada.
Em seguida, vamos avaliar a distribuição pLDDT (Figura 16) ao longo da estrutura. Nota-se que grande parte da estrutura possui um pLDDT > 90, o que demonstra que a estrutura foi modelada com alta confiança.

Figura 16. Visualizando o plDDT ao longo da estrutura.
A ferramenta gera cinco modelos e, na Figura 17, observamos o erro predito ou PAE (Predicted aligned error). Nos eixos x e y encontramos a sequência e, na parte horizontal, temos a divisão de cadeias A, B, C e D. Essa métrica varia de 0 a 30 e, quanto menor o resultado, com mais confiança o complexo foi modelado. Como podemos observar na figura abaixo, a estrutura foi bem modelada, uma vez que o erro predito é baixo em todas as estruturas, tanto da sequência das cadeias quanto das regiões de ligação entre elas.
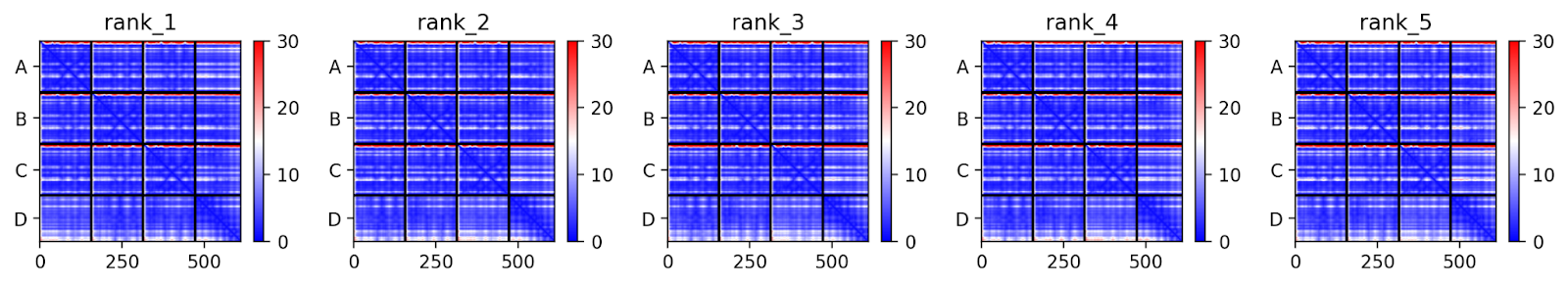
Figura 17. Visualizando o erro predito.
Por fim, podemos visualizar o gráfico de IDDT (Figura 18) por resíduo para os cinco modelos gerados. Nele, observa-se que os cinco modelos tiveram seus resíduos avaliados com IDDT acima de 70, com algumas regiões com IDDT acima de 80. Nota-se que o complexo foi bem modelado, o que já era esperado, uma vez que foram encontrados moldes com alta cobertura.
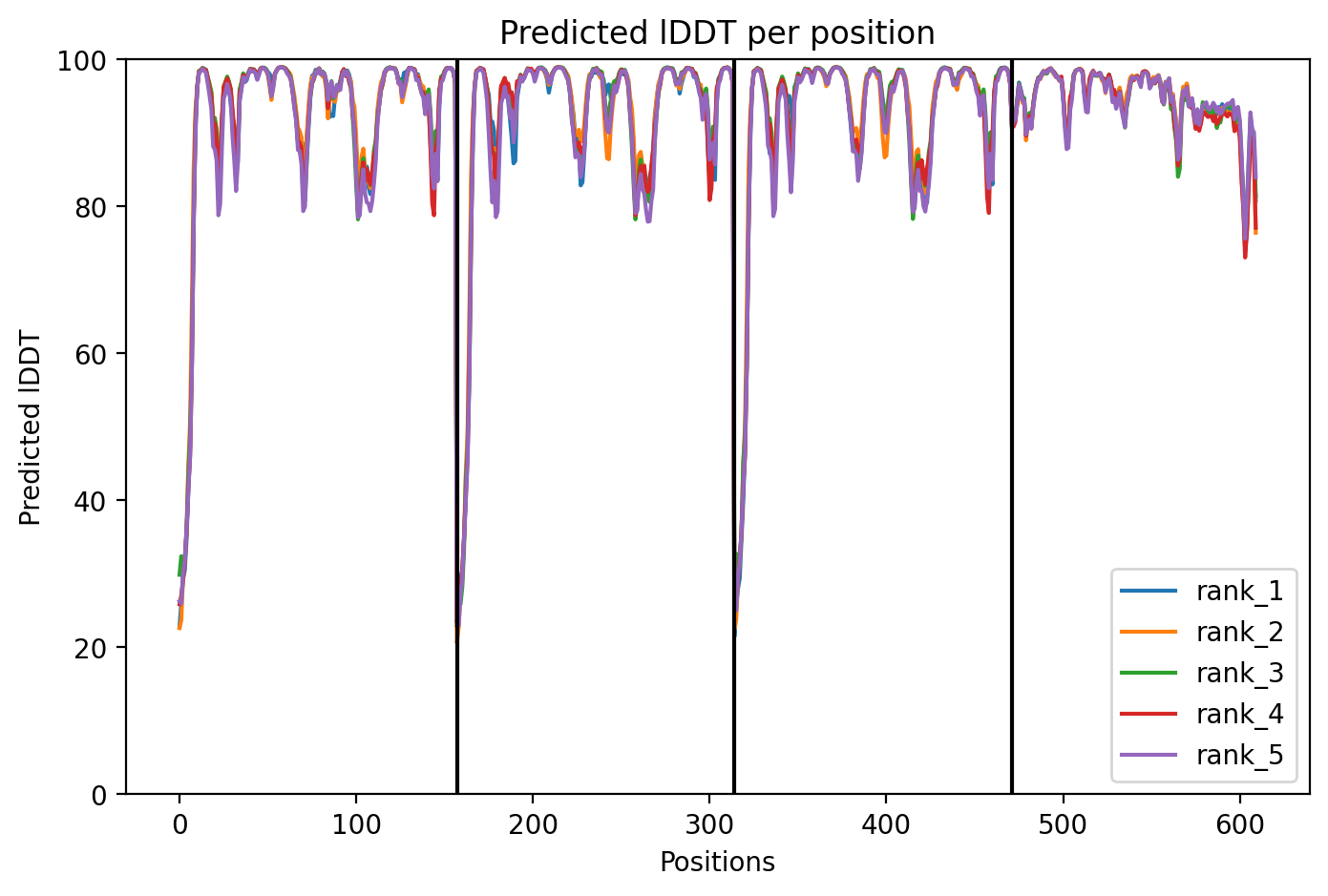
Figura 18. IDDT predito para os cinco modelos gerados.
Conclusão
Por fim, é importante observar que a ferramenta apresenta algumas limitações:
O AlphaFold não é recomendado para análise mutacional, uma vez que utiliza dados já existentes para a construção dos modelos estruturais e, portanto, sua previsão terá uma baixa confiança.
Os modelos construídos não consideram os ligantes: a ferramenta prevê apenas a cadeia peptídica principal, não as estruturas de cofatores, metais e modificações co- e pós-traducionais. Por outro lado, como o modelo é treinado a partir de modelos PDB, muitas vezes com essas modificações anexadas, a estrutura prevista é frequentemente consistente com a estrutura esperada na presença de íons ou cofatores” [16].
As proteínas flexíveis não são modeladas com alta qualidade. Pedaços flexíveis, como regiões C e N-terminais, possuem baixa qualidade.
Apesar das limitações citadas, como vimos neste artigo, o AlphaFold é uma ferramenta revolucionária no que diz respeito à previsão de estruturas de proteínas e tem potencial para melhorar ainda mais à medida que for treinada com mais estruturas. Além disso, é importante ressaltar que essa ferramenta pode ser utilizada para diversos estudos de alto impacto, uma vez que prevê estruturas até então desconhecidas. Dentre eles: desenvolvimento de fármacos, estudo de variantes patogênicas, auxiliar no entendimento de algumas doenças e no desenvolvimento de vacinas [17].
Referências
[1] AlphaFold. Disponível em: https://alphafold.ebi.ac.uk/. Acesso em: 31 de julho de 2023.
[2] Protein Data Bank. Disponível em: https://www.rcsb.org/. Acesso em: 31 de julho de 2023.
[3] AlQuraishi, M. AlphaFold at CASP13, Bioinformatics, Volume 35, Issue 22, November 2019, Pages 4862–4865, https://doi.org/10.1093/bioinformatics/btz422.
[4] AlQuraishi, M. AlphaFold @ CASP13: “What just happened?. Some Thoughts on a Mysterious Universe. Disponível em: https://moalquraishi.wordpress.com/2018/12/09/alphafold-casp13-what-just-happened/. Acesso em: 31 de julho de 2023.
[5] Ramachandran, G. N., Ramakrishnan, C., & Sasisekharan, V. (1963). Stereochemistry of polypeptide chain configurations. Journal of Molecular Biology, 7, 95–99. https://doi.org/10.1016/s0022-2836(63)80023-6.
[6] Branden, C. I., & Tooze, J. (1999). Introduction to Protein Structure (2nd ed.). Garland Science. Disponível em: https://www.routledge.com/Introduction-to-Protein-Structure/Branden-Tooze/p/book/9780815323051.
[7] Fang, C., Shang, Y., & Xu, D. (2018). Prediction of Protein Backbone Torsion Angles Using Deep Residual Inception Neural Networks. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 10.1109/TCBB.2018.2814586. https://doi.org/10.1109/TCBB.2018.2814586.
[8] John Jumper et al., “AlphaFold 2”. Apresentação na CASP 14. Dez 2020. Disponível em: https://predictioncenter.org/casp14/doc/presentations/2020_12_01_TS_predictor_AlphaFold2.pdf.
[9] Jumper, J, Evans, R, Pritzel, A, et al. Applying and improving AlphaFold at CASP14. Proteins. 2021; 89(12): 1711- 1721. doi:10.1002/prot.26257.
[10] Jumper, J. et al., conference abstract (December 2020).
[11] Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). https://doi.org/10.1038/s41586-021-03819-2.
[12] Mirdita, M., Schütze, K., Moriwaki, Y. et al. ColabFold: making protein folding accessible to all. Nat Methods 19, 679–682 (2022). https://doi.org/10.1038/s41592-022-01488-1.
[13] Nature. ‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures. Disponível em: https://www.nature.com/articles/d41586-020-03348-4. Acesso em: 29 ago. 2023.
[14] Eck, M. J., and Sprang, S. R. “The structure of tumor necrosis factor-α at 2.6 Å resolution: Implications for receptor binding.” Journal of Biological Chemistry.
[15] Banner D. W. et al. Crystal structure of the soluble human 55 kd TNF receptor-human TNF beta complex: implications for TNF receptor activation. Cell. 1993 May 7;73(3):431-45. doi: 10.1016/0092-8674(93)90132-a. PMID: 8387891.
[16] O AlphaFold e o desenvolvimento de vacinas. OnlineBioinfo – Comunicação científica em Bioinformática. Disponível em: https://onlinebioinfo.com/2022/02/15/o-alphafold-e-o-desenvolvimento-de-vacinas/. Acesso em: 31 de julho de 2023.
[17] Thornton, J.M., Laskowski, R.A. & Borkakoti, N. AlphaFold heralds a data-driven revolution in biology and medicine. Nat Med 27, 1666–1669 (2021). https://doi.org/10.1038/s41591-021-01533-0.
[18] Mariano, D. AlphaFold e a busca pelo Santo Graal da Biologia Molecular. Revista Brasileira de Bioinformática 1, 162-167 (2022). Disponível em: https://bioinfo.com.br/alphafold-e-a-busca-pelo-santo-graal-da-biologia-molecular.
[19] Finkelstein, A.; Finkelstein, A.V. Protein Folding: Enigma and Solution. Encyclopedia. Available online: https://encyclopedia.pub/entry/8524 (accessed on 15 November 2022); https://medium.com/turing-talks/alphafold-2-entenda-seu-funcionamento-e-implica%C3%A7%C3%B5es-para-biologia-computacional-2ace80b1b70b. Acesso em: 15 de Novembro de 2022.
[20] Thornton, J.M., Laskowski, R.A. & Borkakoti, N. AlphaFold heralds a data-driven revolution in biology and medicine. Nat Med 27, 1666–1669 (2021). https://doi.org/10.1038/s41591-021-01533-0.
Autores: Vivian Morais Paixão https://orcid.org/0000-0002-4154-304X, Angie Atoche Puelles https://orcid.org/0000-0001-8471-8172, Eduardo Utsch Madureira Moreira https://orcid.org/0000-0002-9513-2342, Luana Luiza Bastos https://orcid.org/0000-0002-6932-0438, Raquel Cardoso de Melo-Minardi https://orcid.org/0000-0001-5190-100X
Revisão: Ana Carolina Silva Bulla https://orcid.org/0000-0003-4118-294X, Ariany Rosa Gonçalves https://orcid.org/0000-0001-6887-5362 e Filipe Augusto Teixeira https://orcid.org/0000-0001-9398-1298
Cite este artigo:
Paixão, VM et al. AlphaFold 2: revolucionando a modelagem de estruturas 3D de macromoléculas. BIOINFO. ISSN: 2764-8273. Vol. 3. p.23 (2023). doi: 10.51780/bioinfo- 03-23
[…] AlphaFold 2: revolucionando a modelagem de estruturas 3D de macromoléculas […]
[…] AlphaFold 2: revolucionando a modelagem de estruturas 3D de macromoléculas […]