

A bioinformática é uma área relativamente nova na intersecção da biologia e ciência da computação. Ainda assim, trouxe consigo diversos avanços tecnológicos que atualmente são considerados imprescindíveis para o meio científico mundial [1]. Podemos citar a exemplo as diversas atualizações das tecnologias de sequenciamento que ocorreram somente nas últimas duas décadas [2]. Todavia, sendo uma área do saber recente, a bioinformática quanto à ciência ainda possui pontos de melhoria. Nesse contexto, um tópico que sempre está em voga é a pouca padronização da anotação genômica [3].
O processo de anotação é essencial para o desenvolvimento de metodologias cuja base parte da análise do material genético, tais como pan-genômica e taxogenômica. Em suma, essa etapa da genômica comparativa consiste em dar sentido à sequências biológicas, indicando pontos relacionados aos padrões de presença gênica, possíveis produtos e possíveis funções [4]. Contudo, esse é um passo que, acima de qualquer outro ponto, é dependente de comparações contra bancos de dados. Afinal, para se conhecer o produto que melhor deriva de uma sequência, é preciso comparar a sequência em questão contra outras possíveis candidatas já estudadas [5]. Porém, em bancos de dados imensos e não-curados, sequências idênticas podem possuir diferentes nomes de produto e estarem relacionadas a diferentes genes, ou seja, duas sequencias iguais recebem por vezes nomes não-sinônimos que confundem as ferramentas de predição de contaminam os bancos de dados com informações equivocadas. Outra questão é a presença de genes ou proteínas fragmentadas em bancos de dados utilizados para anotação. Utilizar dados dessa qualidade diminui a acurácia da anotação, já que por vezes pode alterar a fase de leitura ou levar a predição de pseudogenes [6-7].
Pela lógica então, o problema da padronização da anotação genômica deriva diretamente da ausência de padronização dos bancos de dados utilizados para essa função [5-8].
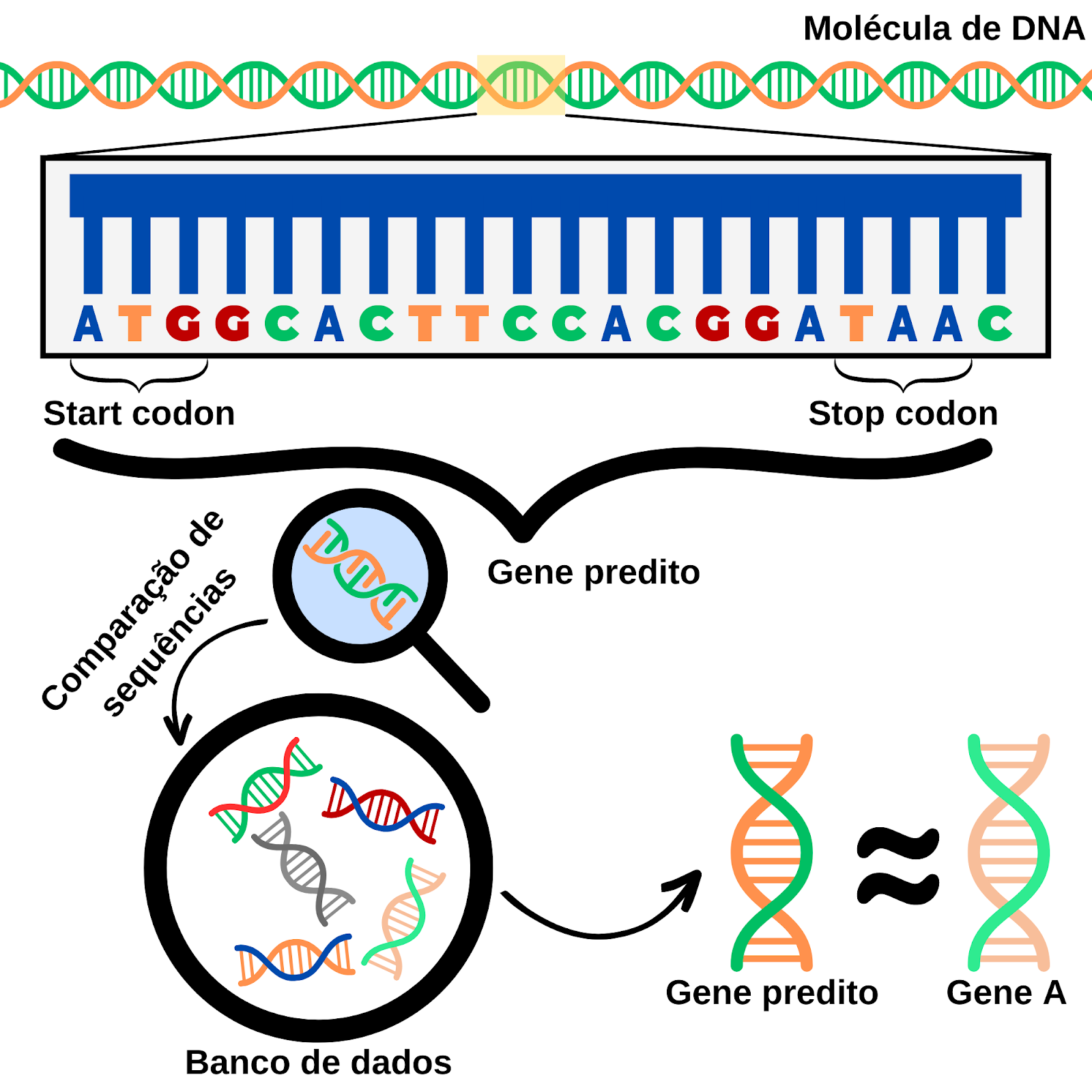 Figura 1: Simplificação gráfica do processo de anotação gênica em um genoma bacteriano. Fonte: próprio autor.
Figura 1: Simplificação gráfica do processo de anotação gênica em um genoma bacteriano. Fonte: próprio autor.
Não obstante, existem alternativas que contornam o problema. Caso apenas uma anotação simples seja necessária, é possível realizar uma anotação automática de todo um dataset de genomas por meio de uma mesma ferramenta ou pipeline. Esse passo visa padronizar os meios de comparação entre as sequências incluídas na análise, garantindo que todas foram comparadas pelo mesmo método e contra as mesmas referências. Porém, essa opção culmina em um passo adicional no pipeline de genômica comparativa, que por vezes é por si só extenso, e pode ser custoso temporalmente a depender do dataset inicial. Ao utilizar uma mesma ferramenta para a comparação e o mesmo banco de dados biológicos, espera-se que sequências similares sejam computadas pela mesma métrica e possuam a mesma terminologia e nomenclatura [7]. Tal fato implica em uma melhor acurácia do resultado final, e permite que o analista trabalhe com um dado limpo e não redundante
Outra alternativa é utilizar anotações públicas de um mesmo banco de dados genômico. Com a criação do banco RefSeq, houve um aumento na qualidade da anotação de genomas disponíveis na plataforma National Center for Biotechnology Information (NCBI) – um dos maiores repositórios de genes e genomas do mundo [9-10]. Essa melhora nas características gerais de anotações públicas do NCBI se deve à uma curadoria fina dos dados depositados globalmente pelos desenvolvedores e envolvidos com a plataforma. A utilização de dados curados aumenta o número de proteínas hipotéticas preditas ao final do processo de anotação, porém eleva a qualidade dos produtos anotados pois garante sua veracidade. Esse foi um passo importante para melhorar a reprodutibilidade de trabalhos genômicos, todavia, essa padronização ainda carece de curadoria por parte daqueles que obtêm esses dados [11].
Portanto, cabe ao bioinformata ou simpatizante da bioinformática a tarefa de selecionar o melhor banco de dados para realizar o processo de anotação, além de desenvolver métodos que solucionem o problema a longo prazo. Vale ressaltar que a bioinformática é uma potência científica, tendo como proposta auxiliar na busca pela solução de problemas biológicos complexos, sendo portanto a área mais adequada para lidar com essa problemática relacionada a ninguém menos que ela mesma.
Referências
[1] Bayat A 2002 Science, medicine, and the future: Bioinformatics BMJ : British Medical Journal 324 1018. DOI: 10.1136/bmj.324.7344.1018
[2] Meera Krishna B, Khan M A and Khan S T 2019 Next-Generation Sequencing (NGS) Platforms: An Exciting Era of Genome Sequence Analysis Microbial Genomics in Sustainable Agroecosystems: Volume 2 ed V Tripathi, P Kumar, P Tripathi, A Kishore and M Kamle (Singapore: Springer) pp 89–109. DOI: https://doi.org/10.1007/978-981-32-9860-6_6
[3] Salzberg S L 2019 Next-generation genome annotation: we still struggle to get it right Genome Biology 20 92. DOI: 10.1186/s13059-019-1715-2
[4] Dominguez Del Angel V, Hjerde E, Sterck L, Capella-Gutierrez S, Notredame C, Vinnere Pettersson O, Amselem J, Bouri L, Bocs S, Klopp C, Gibrat J-F, Vlasova A, Leskosek B L, Soler L, Binzer-Panchal M and Lantz H 2018 Ten steps to get started in Genome Assembly and Annotation F1000Res 7 ELIXIR-148. DOI: 10.12688/f1000research.13598.1
[5] Médigue C and Moszer I 2007 Annotation, comparison and databases for hundreds of bacterial genomes Research in Microbiology 158 724–36. DOI: 10.1016/j.resmic.2007.09.009
[6] Costa M A, Guterres A. A Bioinformática na busca implacável: Onde estão os pseudogenes?. In: BIOINFO – Revista Brasileira de Bioinformática.. Edição #01. Julho, 2022. Acesso: https://bioinfo.com.br/a-bioinformatica-na-busca-implacavel-onde-estao-os-pseudogenes/
[7] Klimke W, O’Donovan C, White O, Brister J R, Clark K, Fedorov B, Mizrachi I, Pruitt K D and Tatusova T 2011 Solving the Problem: Genome Annotation Standards before the Data Deluge Stand Genomic Sci 5 168–93. DOI: 10.4056/sigs.2084864
[8] Maia G A, Filho V B, Kawagoe E K, Teixeira Soratto T A, Moreira R S, Grisard E C and Wagner G 2022 AnnotaPipeline: An integrated tool to annotate eukaryotic proteins using multi-omics data Frontiers in Genetics 13. DOI: https://doi.org/10.3389/fgene.2022.1020100
[9] Nasko D J, Koren S, Phillippy A M and Treangen T J 2018 RefSeq database growth influences the accuracy of k-mer-based lowest common ancestor species identification Genome Biol 19 165. DOI: 10.1186/s13059-018-1554-6
[10] O’Leary N A, et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation Nucleic Acids Res 44 D733-745. DOI: 10.1093/nar/gkv1189
[11] McDonnell E, Strasser K and Tsang A (2018). Manual Gene Curation and Functional Annotation Methods Mol Biol 1775 185–208. DOI: 10.1007/978-1-4939-7804-5_16
—
Autor: Diego Lucas Neres Rodrigues https://orcid.org/0000-0003-2812-3072
Revisão: Ana Carolina Bulla; Bibiana Fam
Cite este artigo:
Rodrigues, DLN. Desafios na padronização da anotação genômica. BIOINFO. ISSN: 2764- 8273. Vol. 3. p.05 (2023). doi: 10.51780/bioinfo-03-05
[…] Desafios na padronização da anotação genômica […]
[…] Desafios na padronização da anotação genômica […]