

A bioinformática é fundamental para a análise de dados biológicos, especialmente no estudo de proteínas, sendo uma área em expansão no Brasil. O uso de plataformas gratuitas e de código aberto tem possibilitado a inclusão de análises computacionais nos cursos de ciências biológicas e da saúde. Dada a relevância das proteínas em processos biológicos, o ensino de bioinformática pode potencializar a formação de futuros profissionais, permitindo a predição de estruturas e funções proteicas com recursos computacionais. Este guia apresenta ferramentas online gratuitas para análises bioinformáticas no ensino da estrutura proteica, incentivando os estudantes a realizarem suas próprias análises. As atividades propostas promovem uma compreensão lúdica e eficiente do tema, integrando conceitos de diferentes disciplinas. Com o uso de recursos como o PSIPRED, MODELLER e AlphaFold, os estudantes podem explorar a estrutura e função das proteínas, preparando-se para enfrentar desafios nos campos da pesquisa e biotecnologia.
Autora: Aline Sampaio Cremonesi (ORCID: https://orcid.org/0000-0002-1283-1237)
Revisão: Bibiana, Bruna Espino, Luana Bastos
Introdução
A Bioinformática desempenha um papel importante na análise de grandes volumes de dados biológicos e na compreensão de funções moleculares, especialmente no estudo de proteínas. No Brasil, o campo tem crescido com o uso de plataformas gratuitas e de código aberto, que facilitam a inclusão de análises computacionais em cursos das ciências biológicas e da saúde [1]. Um exemplo desta importância foi o Prêmio Nobel de Química de 2024 que foi concedido a pesquisadores que utilizaram inteligência artificial para determinar a estrutura de proteínas com alta precisão, por meio do AlphaFold. Este programa usa redes neurais profundas para prever estruturas tridimensionais de proteínas com base em sua sequência de aminoácidos com precisão, mesmo em casos com pouca ou nenhuma informação estrutural disponível, revolucionando o estudo de proteínas e abrindo novas possibilidades para o desenvolvimento de fármacos e a pesquisa biomédica [2].
Estudar proteínas é particularmente relevante, já que essas moléculas estão envolvidas em praticamente todos os processos biológicos e, por isso, aprofundar estes estudos desde o ensino superior, pode contribuir com avanços na pesquisa de futuros profissionais. Como experimentos para este tipo de análises costumam ser de alto custo e exigem infraestrutura especializada, a bioinformática pode servir como uma aliada no ensino da estrutura proteica, pois possibilita a predição de estruturas, interações e funções proteicas apenas com recursos computacionais [3]. Muitas destas análises in silico podem ser relacionadas com conceitos adquiridos em diferentes disciplinas do curso como Bioquímica, Biologia Celular, Fisiologia, Biologia Molecular dentre outras, integrando o aprendizado e promovendo o pensamento multidisciplinar que poderá ser um diferencial ao estudante em atividades desenvolvidas no estágio, na Iniciação Científica e na sua carreira profissional e acadêmica.
Por isso, trago aqui um pequeno guia de ferramentas online e análises bioinformáticas gratuitas que podem ser utilizadas no ensino da estrutura de proteínas em qualquer disciplina. Estas análises permitem tornar a compreensão deste tema mais lúdica e eficiente ao relacionar os conceitos com as possíveis aplicações destas análises. As informações aqui apresentadas são apenas um guia e podem ser adaptadas de acordo com as necessidades peculiares de cada turma e docente, podendo, inclusive, ser realizadas em uma única aula, ou divididas em diferentes aulas.
Uma sugestão é que cada estudante possa realizar suas próprias análises de uma proteína específica em um computador como em uma aula prática de bioinformática, ou que ao menos trabalhe em grupos. O importante é que eles mesmos realizem suas análises com o propósito de “aprender fazendo”. Cabe ressaltar que as análises aqui sugeridas podem ser feitas em tablets e celulares, se for o caso de não haver computadores para todos os estudantes.
Obtenção da sequência FASTA
A estrutura primária de uma proteína é a ordem linear dos aminoácidos, codificada diretamente pela sequência de nucleotídeos do RNA mensageiro (mRNA) [4; 5]. No formato FASTA (Figura 1), a sequência é representada como uma série de letras, onde cada uma corresponde a um aminoácido. A primeira linha corresponde a um código de informações da proteína no banco de dados no qual esta proteína foi obtida. Embora seja a forma mais simples de representação, essa sequência carrega todas as informações necessárias para que a proteína adote sua estrutura tridimensional funcional [5; 6; 7]. As análises in silico começam exatamente aqui, com a estrutura primária fornecendo a base para as predições subsequentes que podem ser obtidas em diferentes bancos de dados biológicos, como o NCBI (National Center for Biotechnology Information – https://www.ncbi.nlm.nih.gov/), o KEGG (Kyoto Encyclopedia of Genes and Genomes – https://www.genome.jp/kegg/) e o UniProt (Universal Protein Resource – https://www.uniprot.org/). Sugiro que escolha apenas um banco de dados para trabalhar durante a aula, de forma a padronizar as análises, mas pode ser interessante, se houver tempo, apresentar estes e outros bancos que achar necessário.
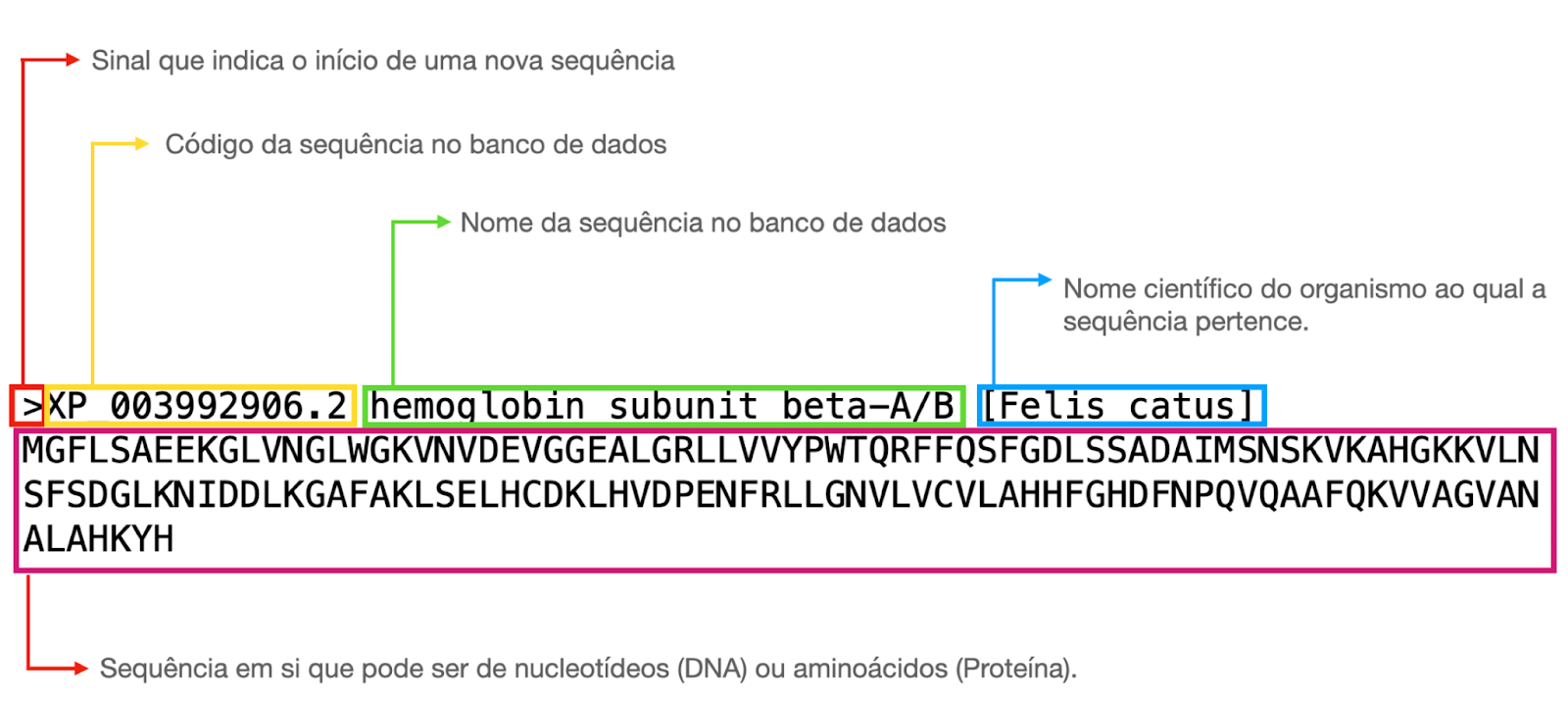 Figura 1: Exemplo de uma sequência FASTA obtida no NCBI. A primeira linha começa com o símbolo “>” que indica onde a sequência começa e, em seguida, há uma série de informações que o próprio banco de dados gera. Ao copiar a sequência FASTA para realizar as análises, pode-se editar a primeira linha, deixando-a mais curta para facilitar a organização e a identificação das sequência, contudo, não pode remover o sinal “>”. A linha de baixo é a sequência de aminoácidos, cada um representado por uma letra específica. Fonte: a própria autora.
Figura 1: Exemplo de uma sequência FASTA obtida no NCBI. A primeira linha começa com o símbolo “>” que indica onde a sequência começa e, em seguida, há uma série de informações que o próprio banco de dados gera. Ao copiar a sequência FASTA para realizar as análises, pode-se editar a primeira linha, deixando-a mais curta para facilitar a organização e a identificação das sequência, contudo, não pode remover o sinal “>”. A linha de baixo é a sequência de aminoácidos, cada um representado por uma letra específica. Fonte: a própria autora.
Dentro do contexto de sala de aula, os estudantes podem escolher uma proteína de interesse ou o docente pode sugerir uma ou mais proteínas para que os estudantes realizem as análises. É importante considerar que proteínas muito grandes (mais de 2000 aminoácidos) ou de membrana podem não ter um bom resultado em todas as análises, por isso, é recomendado que o docente sugira um proteína ou um grupo de proteínas que ele já tenha feito as análises previamente. Proteínas como mioglobina, rodopsina e renina são exemplos que podem ser utilizados em aulas, pois suas sequências FASTA são encontradas facilmente nos bancos de dados mencionados, inclusive de diferentes organismos o que pode ser interessante para comparações nas turmas de cursos que trabalham com diferentes espécies como Ciências Biológicas, Zootecnia e Medicina Veterinária.
Predição de Estrutura Secundária: Hélices-alfa e Folhas-beta
A primeira etapa das análises estruturais é a predição da estrutura secundária da proteína. Ferramentas como o PSIPRED [8; 9] utilizam técnicas baseadas em aprendizado de máquina para identificar padrões de formação de hélices-alfa, folhas-beta e loops (coil). Essas estruturas secundárias são formadas por interações de curto alcance, como ligações de hidrogênio entre os átomos do esqueleto peptídico. A correta identificação dessas características é essencial, pois formam o alicerce sobre o qual a estrutura terciária da proteína se organiza [5].
Esses programas funcionam a partir da inserção da sequência FASTA, que é analisada e comparada com sequências de proteínas cuja estrutura tridimensional já é conhecida. A partir disso, é feito um cálculo de probabilidade para a conformação de cada aminoácido [8; 9]. Os resultados são apresentados em gráficos intuitivos (Figura 2), permitindo que os estudantes compreendam a composição da estrutura secundária da proteína em questão. Com base nesses resultados, os estudantes podem tentar visualizar a estrutura da proteína a partir da composição de estrutura secundária e avaliar quais componentes estão mais presentes: hélice-alfa, folha-beta ou coil.
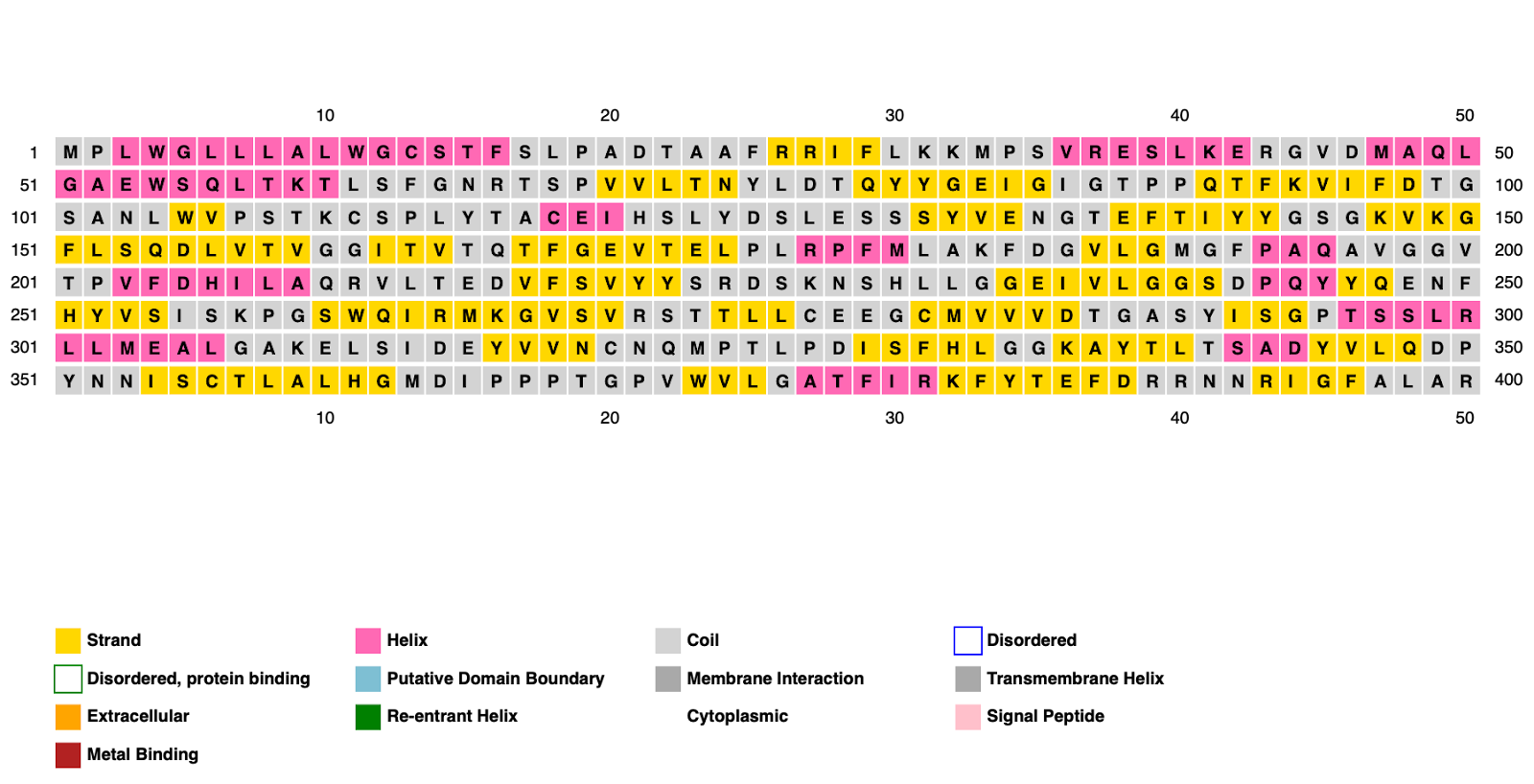
Figura 2: Gráfico gerado pelo Psipred a partir da sequência FASTA. Cada quadrado é um aminoácido indicando a localização de hélices-alfa (em rosa), folhas-beta (em amarelo) e coil (em cinza). Fonte: A própria autora.
Modelagem de Estruturas Proteicas usando Ferramentas Didáticas
A estrutura terciária de uma proteína refere-se ao seu dobramento tridimensional completo, que está diretamente relacionado à sua função biológica. Essa relação é fundamental, uma vez que a forma específica que as proteínas adotam determina como interagem com outras moléculas, como ligantes, DNA e outras proteínas [5; 7]. Compreender essa estrutura permite aos estudantes inferir informações sobre a função da proteína, tornando a modelagem estrutural uma ferramenta aliada ao ensino da estrutura proteica.
Atualmente existem vários programas com diferentes metodologias destinados a gerar modelos tridimensionais de estrutura proteicas a partir da sequência FASTA. Cada método também possui seus próprios mecanismos de validar a qualidade do modelo gerado. Os programas aqui demonstrados estão disponíveis para análises online, sem a necessidade de instalação.
O MODELLER (https://toolkit.tuebingen.mpg.de/tools/hhpred) [10; 11] é amplamente utilizado para a predição da estrutura terciária por meio de métodos de homologia, especialmente quando uma proteína com alta similaridade de sequência já teve sua estrutura resolvida experimentalmente. Neste caso é possível orientar os estudantes a selecionar uma ou mais proteínas-molde (template), fornecendo exemplos práticos de como as similaridades nas sequências de aminoácidos podem resultar em estruturas tridimensionais semelhantes. De forma similar, o SWISS-MODEL (https://swissmodel.expasy.org/) [12] oferece uma interface interativa que pode ser muito útil em ambientes acadêmicos. Ao utilizar essa ferramenta, os estudantes podem facilmente modelar proteínas e visualizar de forma interativa as estruturas previstas. Durante a atividade, os estudantes podem observar como a escolha de um molde apropriado influencia o resultado da modelagem, resgatando conceitos sobre domínios proteicos e conservação de aminoácidos em proteínas com funções similares.
Dentre os resultados (Figura 3) está a estrutura do modelo gerado que pode ser baixado no formato PDB, e também é possível visualizar o diagrama de Ramachandran interativo, que avalia a qualidade do modelo gerado com base nos ângulos phi e psi de cada aminoácido com penalizações para ângulos fora das zonas permitidas [5; 13]. Com base nestas informações é possível discutir em sala de aula sobre a estabilidade estrutural das proteínas e a fidelidade do modelo gerado.
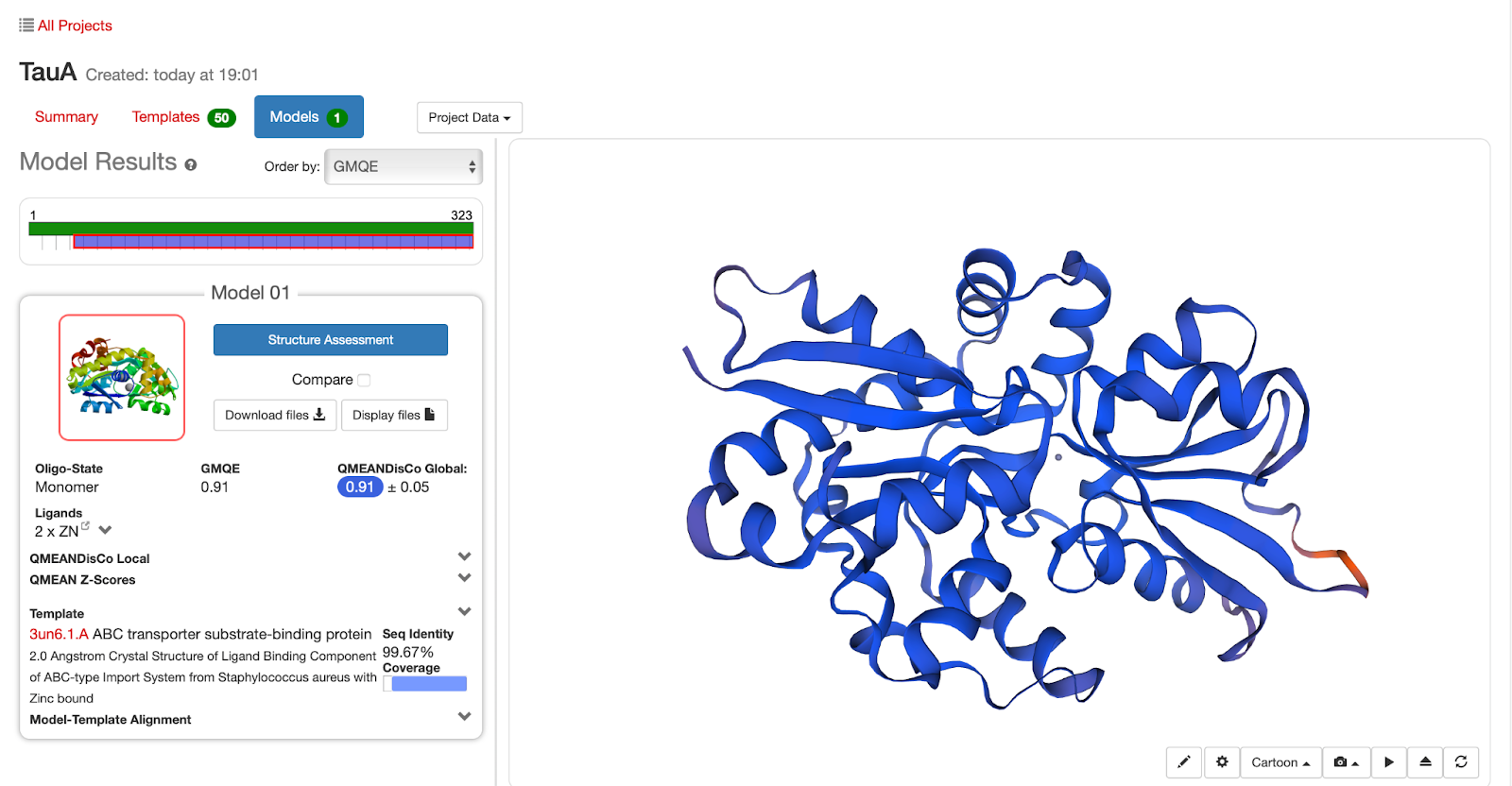 Figura 3: Tela de resultados da modelagem por homologia realizada pelo Swiss Model. O modelo gerado pode ser visualizado de forma interativa à direita da tela enquanto as informações do modelo e do molde se encontram à esquerda. Em “Structure assessment” é possível visualizar o diagrama de Ramachandran referente ao modelo gerado. Fonte: a própria autora.
Figura 3: Tela de resultados da modelagem por homologia realizada pelo Swiss Model. O modelo gerado pode ser visualizado de forma interativa à direita da tela enquanto as informações do modelo e do molde se encontram à esquerda. Em “Structure assessment” é possível visualizar o diagrama de Ramachandran referente ao modelo gerado. Fonte: a própria autora.
Nem todas as proteínas possuem homólogas com estrutura resolvida para realizar uma modelagem por homologia, por isso, o I-TASSER (https://zhanggroup.org/I-TASSER/) [14] é uma alternativa neste caso. Ele combina predições de estrutura baseadas em homologia com a geração de modelos ab initio, permitindo que os estudantes explorem domínios funcionais e predições de interações proteína-proteína. Isto abre espaço para discussões sobre a complexidade das interações biomoleculares e a importância da modelagem na bioinformática.
Por fim, temos o aclamado AlphaFold (https://alphafold.ebi.ac.uk/ ) [2], que revolucionou a predição de estruturas de proteínas ao utilizar inteligência artificial para prever conformações tridimensionais com alta precisão. Ao analisar a sequência FASTA, o AlphaFold usa redes neurais profundas para gerar modelos tridimensionais baseados em padrões aprendidos de dados estruturais conhecidos. Este método é útil para proteínas que não possuem homólogos estruturais conhecidos ou apresentam dificuldades para modelagem, como proteínas de membrana e proteínas longas. A versão inicial do AlphaFold requer hardware potente, como GPUs (Unidades de Processamento Gráfico) de alta performance, o que pode ser um desafio em algumas instituições de ensino. Porém atualmente é possível realizar estas modelagens online pelo AlphaFold Server (https://alphafoldserver.com/about) [15] que é mantido pelo AlphaFold 3 devido a uma colaboração entre o EMBL-EBI e a DeepMind. Ele permite modelar a partir da sequência FASTA, estruturas de proteínas e outras biomoléculas, como DNA e RNA, com precisão de alta resolução entre poucos minutos a algumas horas, dependendo da complexidade da molécula. Os resultados (Figura 4) são apresentados em forma de estrutura tridimensional com escalas de cores para indicar a confiança das previsões. Outro método de usar os recursos AlphaFold é a partir do ColabFold (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb) [16] que surgiu para oferecer uma solução acessível para os usuários realizarem as análises do AlphaFold diretamente no Google Colab, democratizando o acesso à tecnologia avançada além de apresentar uma interface intuitiva e flexibilidade na personalização das predições.
A validação de modelos gerados pelo AlphaFold utiliza métricas como lDDT (Local Distance Difference Test – Teste de Diferença de Distância Local) e PAE (Predicted Aligned Error – Erro Alinhado Previsto). O lDDT (pontuação por átomo de 0 a 1) indica a confiabilidade do modelo onde valores acima de 0,9 representam alta precisão, enquanto valores abaixo de 0,7 indicam baixa confiança. O PAE estima o erro esperado no posicionamento relativo de domínios, comparando distâncias entre resíduos no backbone. Gráficos do ColabFold mostram esses valores com faixas coloridas, onde azul indica alta precisão e vermelho sugere maior erro [2].
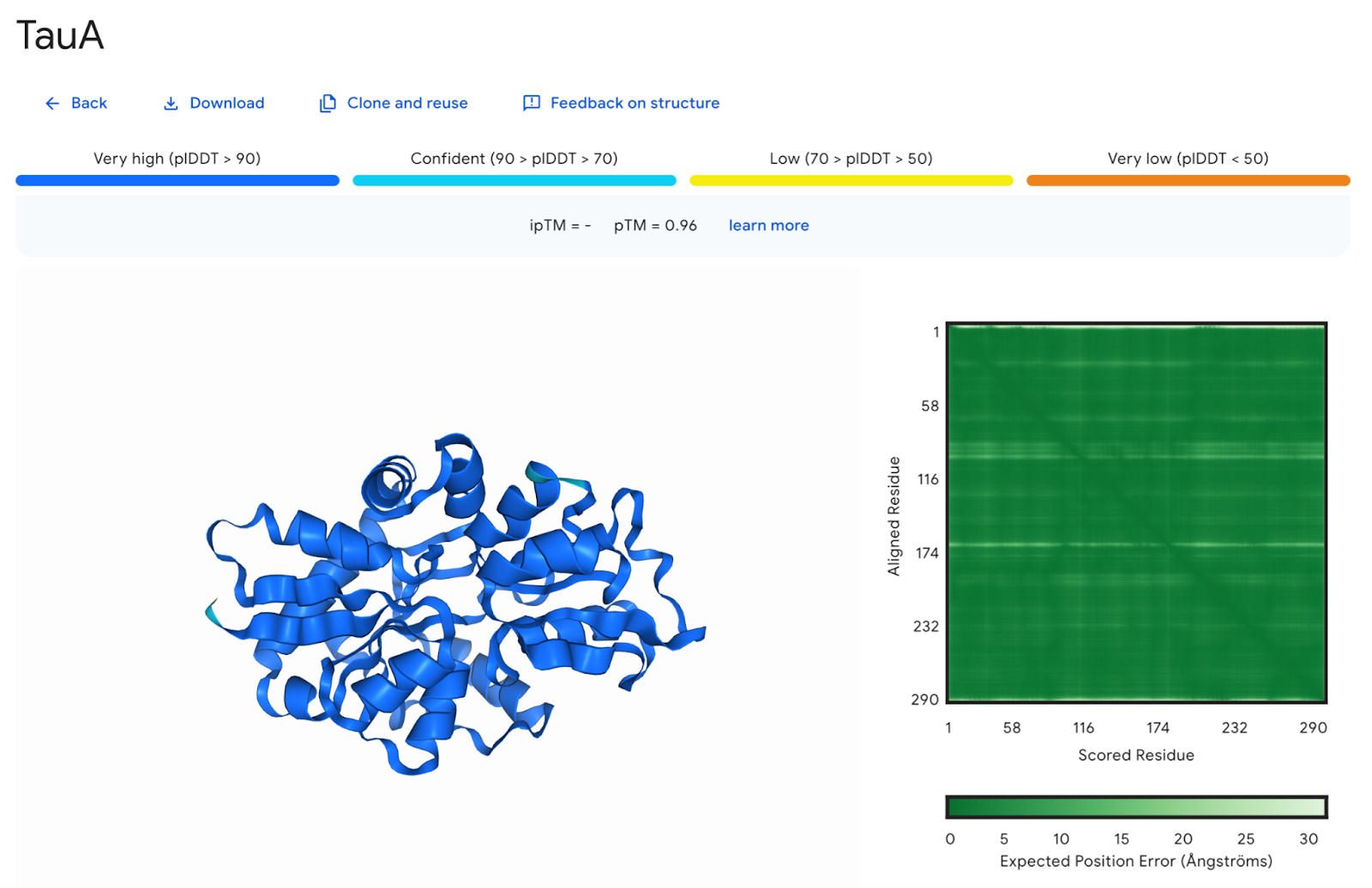 Figura 4: Tela de resultados da modelagem, usando AlphaFold Server. O modelo gerado pode ser visualizado de forma interativa à esquerda da tela, cuja cor é baseada nos valores preditos do Teste de Diferença de Distância Local (plDDT) acima da tela, e o Erro Alinhado Previsto (PAE) do modelo se encontra à direita. Fonte: a própria autora.
Figura 4: Tela de resultados da modelagem, usando AlphaFold Server. O modelo gerado pode ser visualizado de forma interativa à esquerda da tela, cuja cor é baseada nos valores preditos do Teste de Diferença de Distância Local (plDDT) acima da tela, e o Erro Alinhado Previsto (PAE) do modelo se encontra à direita. Fonte: a própria autora.
Visualização da Estrutura Proteica
Todos os programas de modelagem permitem o download do modelo gerado no formato PDB, que requer um software específico para visualização interativa, como o PyMOL [17] ou o Chimera X [18]. Contudo, a instalação de tais programas nos computadores institucionais pode ser complicada em algumas instituições. Nesse contexto, o SWISS-MODEL e o AlphaFold Server se destacam ao permitir a visualização online e interativa dos modelos gerados, favorecendo discussões em sala de aula sobre a estrutura das proteínas e suas relações funcionais.
Para aqueles que optarem por utilizar métodos como o ColabFold, ainda há a possibilidade de visualizar os modelos gerados em um visualizador online, como o Mol 3D Viewer* (https://www.rcsb.org/3d-view) disponível no site do PDB (Protein Data Bank – Banco de Dados de Proteínas). Ao inserir qualquer arquivo PDB, os estudantes podem explorar interativamente as estruturas das proteínas, tornando a aula ainda mais rica e envolvente.
Aplicações: Da Pesquisa à Biotecnologia
A predição de estrutura e função de proteínas tem se consolidado como uma ferramenta essencial na busca por novos alvos terapêuticos, contribuindo significativamente para o avanço no tratamento de diversas doenças e infecções [19; 20]. Por isso, é importante que esse tema seja abordado durante a graduação uma vez que, ao expor os estudantes a essas metodologias, não apenas ampliamos seu entendimento sobre o papel das proteínas na biologia molecular, como também os capacitamos a aplicar, explorar e até mesmo aperfeiçoar essas ferramentas em futuras pesquisas e análises. Esse conhecimento será fundamental para formar profissionais aptos a inovar em áreas como a bioinformática, biotecnologia e saúde, preparando-os para enfrentar desafios científicos com ferramentas modernas e soluções criativas.
As informações aqui apresentadas podem contribuir para o ensino da estrutura de proteínas no Ensino Superior com a ajuda de análises in silico, que também são aplicadas em pesquisa, mas elas podem e devem ser adaptadas de acordo com as necessidades de cada turma, disciplina e curso. O foco é disseminar práticas que podem contribuir com o ensino e divulgar a Bioinformática que é uma área promissora e crescente na ciência, preparando os estudantes para novas oportunidades no presente e no futuro.
Referências
[1] Paixão-Côrtes, V. S. M., et al. A panorama on selection and use of bioinformatics tools in the Brazilian University context. In: Universal Access in Human-Computer Interaction. Virtual, Augmented, and Intelligent Environments: 12th International Conference, UAHCI 2018, Held as Part of HCI International 2018, Las Vegas, NV, USA, July 15-20, 2018, Proceedings, Part II 12. Springer International Publishing, 2018, pp. 553-573. DOI: https://doi.org/10.1007/978-3-319-92052-8_44
[2] Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583-589, 2021. DOI: https://doi.org/10.1038/s41586-021-03819-2
[3] Kann, M. G. Protein interactions and disease: Computational approaches to uncover the etiology of diseases. Briefings in Bioinformatics, 8(5), 333-346, 2007. DOI: https://doi.org/10.1093/bib/bbm031
[4] Lodish, H., Berk, A., Matsudaira, P., Kaiser, C. A., Krieger, M., Scott, M. P., Zipursky, S. L., & Darnell, J. E. (2014). Biologia celular e molecular (7ª ed.). Artmed.
[5] Nelson, D. L., & Cox, M. M. Princípios de bioquímica de Lehninger. Artmed Editora, 2022.
[6] Baker, D., & Sali, A. Protein structure prediction and structural genomics. Science, 294(5540), 93-96, 2001. DOI: https://doi.org/10.1126/science.1065659
[7] Cremonesi, A. S. Bases da bioquímica molecular: Estruturas e processos metabólicos. Editora Intersaberes, 2020.
[8] Buchan, D. W. A., & Jones, D. T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Research, 47, W402-W407, 2019. DOI: https://doi.org/10.1093/nar/gkz297
[9] McGuffin, L. J., Bryson, K., & Jones, D. T. The PSIPRED protein structure prediction server. Bioinformatics, 16(4), 404-405, 2000. DOI: https://doi.org/10.1093/bioinformatics/16.4.404
[10] Zimmermann, L., Stephens, A., Nam, S. Z., Rau, D., Kübler, J., Lozajic, M., Gabler, F., Söding, J., Lupas, A. N., & Alva, V. A completely refactored MPI Bioinformatics Toolkit with a new HHpred server at its core. Journal of Molecular Biology, 430(15), 2237-2243, 2018. DOI: https://doi.org/10.1016/j.jmb.2017.12.007
[11] Gabler, F., Nam, S. Z., Till, S., Mirdita, M., Steinegger, M., Söding, J., Lupas, A. N., & Alva, V. Protein sequence analysis using the MPI Bioinformatics Toolkit. Current Protocols in Bioinformatics, 72(1), e108, 2020. DOI: https://doi.org/10.1002/cpbi.108
[12] Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., Heer, F. T., de Beer, T. A. P., Rempfer, C., Bordoli, L., Lepore, R., & Schwede, T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Research, 46(W1), W296-W303, 2018. DOI: https://doi.org/10.1093/nar/gky427
[13] Sobolev, O. V. et al. A global Ramachandran score identifies protein structures with unlikely stereochemistry. Structure, 28(11), 1249-1258.e2, 2020. DOI: https://doi.org/10.1016/j.str.2020.08.005
[14] Yang, J., & Zhang, Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Research, 43(W1), W174-W181, 2015. DOI: https://doi.org/10.1093/nar/gkv342
[15] Abramson, J., Adler, J., Dunger, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500, 2024. https://doi.org/10.1038/s41586-024-07487-w
[16] Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nature Methods, 19(6), 679-682, 2022. DOI: https://doi.org/10.1038/s41592-022-01488-1 .
[17] DeLano, W. L. PyMOL: An open-source molecular graphics tool. CCP4 Newsletter on Protein Crystallography, 40(1), 82-92, 2002.
[18] Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25(13), 1605-1612, 2004. DOI: https://doi.org/10.1002/jcc.20084.
[19] Li, Z. et al. In silico prediction of drug-target interaction networks based on drug chemical structure and protein sequences. Scientific Reports, 7(1), 11174, 2017. DOI: https://doi.org/10.1038/s41598-017-10724-0.
[20] Macalino, S. J. Y. et al. Evolution of in silico strategies for protein-protein interaction drug discovery. Molecules, 23(8), 1963, 2018. DOI: https://doi.org/10.3390/molecules23081963.