

Revisão:
BIOINFO – Revista Brasileira de Bioinformática. Edição #. .
DOI:
Neste artigo, vamos falar sobre imunobioinformática. Você sabe o que é isso? Já ouviu falar sobre? Pois bem, como o nome sugere a imunobioinformática é sim a junção dos vastos conhecimentos da imunologia com a prática de estratégias de bioinformática. Ou também pode ser entendida como a aplicação das diversas ferramentas e estratégias de bioinformática voltados aos desafios no campo da imunologia. Mas que tipos de desafios? E quais estratégias? É sobre isso que vamos conversar neste capítulo.
Vale lembrar que existem diferenças entre a imunobioinformática e a imunologia computacional. Por imunologia computacional entendemos os trabalhos que visam compreender a imensa complexidade do sistema imune. Sejam através do desenvolvimento de abordagens computacionais poderosas – processando, modelando e integrando grandes volumes de dados imunológicos – ou pela exploração de mecanismos moleculares subjacentes à dinâmica das células imunológicas, através de métodos biológicos de alto rendimento. Já a imunobioinformática está diretamente relacionada com o desenvolvimento de profilaxias contra agentes infecciosos, através da prospecção e da descoberta de candidatos profiláticos, quer sejam estes alvos de droga ou candidatos a vacina, como vamos ver a seguir.
Além disso, aqui nós vamos falar sobre os tópicos de vacinologia reversa, genômica subtrativa e, para explicitar o trabalho da área, vamos fechar com um pequeno estudo de caso, aplicando algumas das técnicas abordadas em imunobioinformática.
Mas antes de começarmos, gostaríamos de relembrar com você, leitor, alguns conceitos básicos de imunologia que serão importantes para a nossa conversa fluir mais adiante. Mas não se preocupe, vamos com bastante calma.
Imunologia básica
O primeiro desses conceitos básicos em imunologia clássica é o dos tipos de imunidade do sistema imune. Mais especificamente, as imunidades inata e adaptativa.
Nós, humanos, temos diversos tipos distintos de proteção contra agentes externos e alheios ao nosso corpo, sejam esses patógenos (i.e., bactérias, vírus, protozoários) ou partículas alergênicas de qualquer natureza, como pólen, glúten, dentre outras.

Para nos proteger dessas várias ameaças e assim evitar possíveis invasões que desequilibrem a saúde do indivíduo, o nosso organismo dispõe de diversas barreiras físicas, químicas e biológicas, como a pele, cílios das pálpebras e sistema respiratório, lágrimas, dentre outras (Figura 1). Entretanto, no caso de uma invasão inevitável, entram em ação as nossas próximas linhas de defesa da saúde: as linhagens celulares do sistema imune [1].
Essas células podem ser de diversos tipos e cumprem distintas funções nesse campo de batalha. Por exemplo, as células de leucócitos, neutrófilos e macrófagos atuam diretamente no combate de agentes infecciosos. Anticorpos produzidos pelos linfócitos do tipo B, ou células do tipo B, também podem atuar bloqueando a ação e eliminando patógenos.
A resposta natural e inespecífica do organismo a ameaças do ambiente é conhecida como ~imunidade inata~, sendo os seus principais mecanismos a fagocitose, a liberação de mediadores inflamatórios e a ativação de proteínas. Contudo, quando ela não é o suficiente para responder às ameaças enfrentadas por um indivíduo, podemos contar com a nossa ~imunidade adaptativa~ [1].
A imunidade adaptativa tem como propósito o de evoluir as defesas de um organismo, através da exposição e resposta específica a estímulos de agentes externos, causadores de doenças ou não. Essa imunidade, também conhecida como adquirida, depende da ativação de células especializadas, chamadas de linfócitos. As principais classes de linfócitos são os linfócitos do tipo B e do tipo T. Eles são responsáveis por mediar os dois tipos de imunidade adquirida que possuímos: as imunidades humoral e celular (Figura 2).

A imunidade humoral se dá pelo reconhecimento específico de antígenos pelos chamados anticorpos, processo que é mediado pelos linfócitos B. Já a imunidade celular é o nosso mecanismo de defesa específico que é mediado por células, os linfócitos T. Os linfócitos T se subdividem em células T auxiliares (T helper) e células T citotóxicas (T cytotoxic), chamados também de células TCD4+ e TCD8+, respectivamente (Figura 2). A importância dessas distintas linhagens de célula T se deve pelos distintos tipos de infecção combatidos por elas. Enquanto as células TCD4+ combatem agentes estranhos dispersos no meio extracelular, como vírus, toxinas e bactérias, as células TCD8+ agem contra perturbações intracelulares, seja contra células tumorais ou infectadas por vírus, por parasitas unicelulares, dentre outros [2].
Você sabia?
As células B e T possuem esse nome especial em função dos principais órgãos produtores desses tipos de linfócitos! Enquanto os linfócitos T são predominantemente produzidos no Timo, os linfócitos B são produzidos pela medula óssea – do inglês, Bone narrow.
Embora alguns antígenos possam estimular diretamente a resposta imunitária, as células T precisam de um mediador para realizar o seu reconhecimento, a célula apresentadora de antígenos (APC). Como os linfócitos T não podem se ligar ao antígeno diretamente, ele precisa ter os peptídeos decompostos do antígeno apresentados a ele por uma APC. Essa “apresentação” acontece pela junção dos receptores de célula T (TCR) com os complexos principais de histocompatibilidade (MHC), moléculas presentes na superfície celular de linhagens celulares que podem atuar como APC, tais como macrófagos, células dendríticas, células de Langerhans e de Kupffer. E é no interior das moléculas do complexo MHC que os peptídeos dos antígenos decompostos são expostos (Figura 3) [2,3].
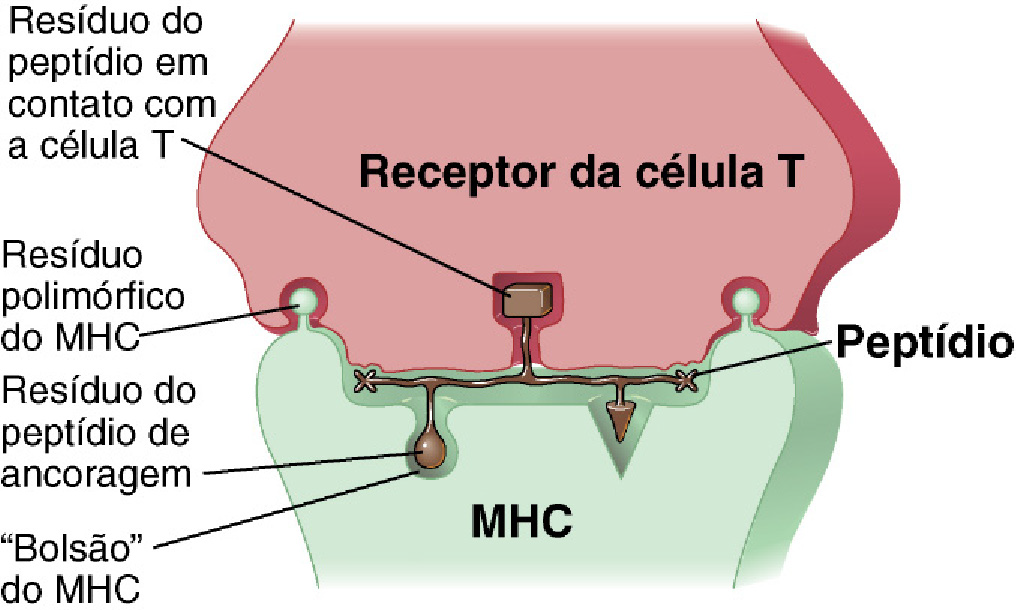
Existem dois tipos de MHC: MHC classe I e MHC classe II. MHC classe I se apresenta a células T citotóxicas, TCD8+, e MHC classe II se apresenta às células T auxiliares, TCD4+. Os termos “CD4” e “CD8” positivos fazem referência a expressão na superfície dos linfócitos T dessas moléculas que atuam como correceptores específicos de MHC classe II e I, respectivamente. Essas moléculas de MHC II e I são altamente variáveis, os genes que as expressam são extraordinariamente polimórficos, sendo essa uma das maiores razões para a diversidade do sistema imune e sua capacidade de responder e se adaptar a ameaças nunca vistas antes pelo organismo [4].
Além disso, um último detalhe importante acerca desse reconhecimento é que o reconhecimento molecular não se dá pelo contato com a estrutura completa dessas estruturas proteicas, apenas uma mínima porção dessas moléculas entra em contato direto com os receptores. O quê? Mas como assim? Vamos explicar.
Essas frações mínimas se chamam ~epítopos~. Epítopos são curtas sequências de aminoácidos (AAs), sendo os fragmentos de menor tamanho possível capazes de induzir a sua identificação, seja por receptores ou por anticorpos. Esses fragmentos não são necessariamente lineares, ou seja, reconhecidos em sua estrutura primária de AAs. Quando uma forma tridimensional específica de estrutura proteica é necessária para o reconhecimento, chamamos essa sequência mínima de epítopo conformacional (Figura 4). Por conta dessa flexibilidade estrutural de sequências de resíduos, o tamanho de epítopos costuma variar dentre 8 e 14 AA, sendo mais comum fragmentos de 9 resíduos de AA (9-mer) [5].

Ufa, agora sim! Acabamos de revisar aspectos básicos em imunologia e estamos prontos para embarcar nos tópicos de imunobioinformática! O próximo tópico se chama Vacinologia Reversa, estratégia voltada para prospecção de candidatos à vacina para o combate de doenças infecciosas. Apesar do que o nome indica, os princípios de RV também auxiliam na busca por alvos para o diagnóstico de doenças.
Você está pronto? Vamos lá!
Vacinologia Reversa
Para começar: as vacinas representam a estratégia profilática mais eficaz na história da medicina para o controle da propagação de doenças infecciosas – aumentando sem precedentes a expectativa de vida humana. Desde o seu advento, a erradicação da varíola e a redução massiva de outras doenças infecciosas, como a poliomielite, o sarampo e a difteria, foram algumas das principais conquistas em saúde pública do século passado, todas alcançadas através da vacinação [6].
Você sabia?
Os termos “vacina” e “vacinação” surgiram em 1796, com os estudos de Edward Jenner acerca do desenvolvimento de vacinas contra a varíola, evitando a infecção ao se isolar materiais a partir da varíola bovina para imunização de pacientes. Do latim vaccinus, que significa ‘derivado da vaca’.
Com base nos princípios de Pasteur, “isolar, inativar e injetar”, os métodos tradicionais utilizados em vacinas de primeira e segunda geração concentraram-se em organismos inteiros, marcando o início do desenvolvimento de vacinas [7]. Com a disponibilidade do sequenciamento de genoma completo do final do século XX, a predição computacional de antígenos assumiu o foco dos estudos para o desenvolvimento de vacinas de terceira geração [8].
Em especial, com o auxílio da bioinformática e o advento da vacinologia reversa (Reverse Vaccinology – RV). Por exemplo, o primeiro trabalho que aplicou a abordagem de RV, contra a bactéria Neisseria meningitidis sorogrupo B (MenB), levou menos de 18 meses para identificar mais vacinas candidatas em MenB do que foi descoberto durante os últimos 40 anos por métodos convencionais, ou seja, isso acelerou drasticamente o processo de desenvolvimento da vacina (Figura 5) [9].

O termo “reversa” de RV faz referência ao caminho de descoberta invertido usado para busca de novas vacinas. Mas, como assim? Bem, na vacinologia clássica, os imunizantes eram desenvolvidos a partir do organismo inteiro, com vacinas a base de patógenos inteiros, enfraquecidos ou quebrados em fragmentos. Nesse sentido, a RV faz o caminho contrário – buscando dentro do(s) genoma(s) por possíveis proteínas, ou partes de proteínas, que, como um conjunto mínimo antigênico, sejam capazes de elicitar resposta imune adaptativa específica contra esse mesmo patógeno [11].
Em suma, a vacinologia reversa analisa a sequência genômica de um patógeno, partindo do genoma completo ao invés do organismo inteiro, e identificando todo o seu catálogo de proteínas com potencial de serem expressas pelo microrganismo a qualquer momento. Esta estratégia tem também a vantagem de ser aplicável tanto a espécies cultiváveis quanto a não cultiváveis [12].
De modo geral, em estudos de RV, as Open Reading Frames (ORFs) derivadas da sequência de um genoma são analisados por um conjunto de softwares (como por exemplo, Vaxign2 [13], NetMHC 4.0 [14], MED. 1.0 [5]) para aferir e predizer atributos desejáveis dos prováveis produtos gênicos das ORFs como candidatos à vacina (Figura 6). Em especial, em atenção às proteínas exportadas, pois estas são essenciais nas interações hospedeiro-patógeno, tais como: adesão às células hospedeiras; invasão intracelular; danos teciduais; resistência ao estresse ambiental da maquinaria de defesa; e mecanismos para subversão da resposta imune do hospedeiro [15–18].

Fatores como localização subcelular e o número de domínios transmembranares são frequentemente considerados na filtragem bioinformática para um alvo vacinal, uma vez que proteínas da membrana externa contendo mais de uma hélice transmembrana são, em geral, difíceis de clonar e purificar [20]. Com a disponibilidade de cepas não patogênicas para diversas espécies, elementos exclusivos de genomas de linhagens virulentas também começaram a ser avaliados em estudos de RV [21]. Além disso, os alvos de vacina preditos são analisados quanto à similaridade de sequência com proteínas do hospedeiro, seja este humano ou animal, a fim de evitar a ineficácia de candidatos à vacina por resistência autoimune [22].
Desde a concepção da RV, o progresso na análise genômica, proteômica e transcriptômica surtiu um enorme impacto no modo com que novos antígenos estão sendo identificados. Para otimizar vacinas baseadas em epítopos, tornou-se uma tarefa essencial prever epítopos imunológicos com base na estrutura de antígenos protetores. Avanços nas tecnologias de clonagem de linfócitos B permitiram a geração de anticorpos monoclonais humanos a partir de células B de memória e plasmablastos, permitindo a construção de bancos de dados dessas estruturas e, assim, a triagem de alvos contra anticorpos humanos e uma melhor caracterização da imunogenicidade dos alvos selecionados [23].
Além disso, ao invés de buscar por candidatos à vacina em uma única linhagem de um patógeno, é possível realizar a prospecção em inúmeros genomas coletivamente, explorando possíveis antígenos para toda a espécie. A disponibilidade de grande número de genomas nos bancos de dados públicos levou ao surgimento da RV Pangenômica (PGRV), ou PanRV (Figura 7). A PanRV aplica os conceitos de genomas core, acessório e unique das ciências pangenômicas na pesquisa em alvos vacinais. Do ponto de vista da vacina, os genomas core e unique são os melhores candidatos para compor uma vacina que seja adequada para todas as cepas estudadas, sem perder de vista as particularidades de cada estirpe [24].

Desde o primeiro trabalho em RV para o desenvolvimento de uma vacina, o conceito de vacinologia reversa também foi aplicado com sucesso a diversos outros agentes de doenças infecciosas. Dentre eles, os vírus Ebola [25] e Zika [26], as bactérias Streptococcus pneumoniae [27] e Mycoplasma pneumoniae [28], além de outros patógenos de interesse veterinário e zoonótico, como Corynebacterium pseudotuberculosis [29] e Brucella spp. [30]. Quanto às ISTs, esforços contínuos estão sendo feitos por pesquisadores para identificar novos candidatos a vacinas contra várias infecções sexualmente transmissíveis, como as causadas por Herpes Simplex Virus-1 [31], Chlamydia trachomatis, clamídia [32], e Treponema pallidum, sífilis [33].
Enormes avanços em pesquisa na busca por profiláticos foram alcançados com o auxílio da vacinologia reversa! Mas nem tudo são vacinas… o próximo tópico em imunobioinformática traz as contribuições da genômica subtrativa para a prospecção de alvos de drogas e a descoberta de novos alvos farmacológicos.
Vamos lá!
Genômica Subtrativa
Para você, leitor, entender a necessidade de que se desenvolvam novas drogas: no ano de 2020, a Organização Mundial da Saúde (OMS) declarou que a resistência antimicrobiana (AMR) é uma das 10 principais ameaças à saúde pública e ao desenvolvimento global enfrentadas pela humanidade no séc. 21. Apenas na União Europeia e EUA, a resistência a antibióticos é responsável por aproximadamente 50.000 óbitos anuais [34].
Além disso, vários dos medicamentos utilizados pela medicina atual para o tratamento de agentes infecciosos causam efeitos colaterais de pequeno a grande porte, o que também compromete a qualidade de vida dos pacientes. Todos esses fatores somados impõem a necessidade de se identificar drogas novas e mais eficazes para o combate de doenças [35].
Os primeiros projetos de sucesso para o design racional de drogas foram publicados há não muito tempo, apenas no início dos anos 1990. Contudo, atualmente, o desenho de medicamentos baseados em sua estrutura molecular é uma parte vital da maioria dos programas industriais de descoberta de medicamentos e, também, um importante campo da pesquisa acadêmica [36].
Nesse contexto, a abordagem chamada de ~genômica subtrativa~ está amplamente envolvida na identificação de novos e específicos alvos de drogas contra organismos patogênicos, como um passo para o reposicionamento ou desenvolvimento de novos fármacos [37].
A genômica subtrativa faz alusão a abordagem matemática, onde “subtração” significa literalmente “removido de baixo”, ou seja, retirar um pedaço menor de um maior. Geralmente, dois ou mais genomas são utilizados para determinar a diferença entre as informações analisadas e o conjunto de dados genômicos são subtraídos uns dos outros a fim de revelar os genes específicos de gênero, espécie e fenótipo único [38,39].
A identificação de alvos em genômica subtrativa é majoritariamente baseada em genes essenciais e não-homólogos ao hospedeiro. Genes essenciais são genes necessários para o crescimento, adaptabilidade e sobrevivência de um organismo, sendo letal a deficiência de qualquer um desses genes para o organismo. Nesse sentido, o Banco de Dados de Genes Essenciais (DEG) [40] é o principal repositório que lista genes essenciais validados experimentalmente em bactérias, fungos, plantas e animais.
O DEG é comumente empregado para a identificação de alvos por abordagens genômicas subtrativas. Um gene não-homólogo de um patógeno seria aquele que não está presente no hospedeiro, mas está presente no patógeno, sendo este considerado o tipo ideal de alvo contra um agente infeccioso [41].
Por vias de praxe, um alvo de drogas proteico deve atender a quatro critérios principais:
- deve ser um gene essencial para a sobrevivência ou patogênese do organismo alvo;
- “drogabilidade”, i.e., possuir características estruturais de proteína que a tornem capaz de se ligar a pequenas moléculas inibidoras;
- caracterização funcional e estrutural, com ensaios estabelecidos para testar a inibição por pequenas moléculas;
- distinção de alvos de drogas atuais para evitar resistência cruzada.
Além desses princípios, alguns trabalhos em genômica subtrativa levam em consideração algumas outras estratégias para refinar ainda mais a sua triagem por alvos de drogas. Inúmeros trabalhos avaliam, também, a predição da localização subcelular de suas proteínas pré-selecionadas, pois, por estarem mais expostas e voltadas para o meio extracelular, proteínas de membrana são consideradas excelentes alvos de drogas [42,43].
Além disso, somados à genômica subtrativa, estudos computacionais de acoplamento ou docking molecular podem levar à descoberta de novos medicamentos para o tratamento de infecções [37]. Na triagem virtual, pequenas moléculas ou compostos são encaixados computacionalmente na região de interesse do alvo e classificados com base nas interações mais favoráveis, preditas contra o sítio-ativo de ligação da proteína alvo (Figura 8).
Existem diversos bancos de dados de biomoléculas para docking, e.g., ZINC Database [44], Available Chemicals Database (ACD) [45]. A principal vantagem de realizar o docking molecular com compostos de bancos como esses é que os ligantes de sucesso podem ser adquiridos e, com maior praticidade, levados para ensaios in vitro e testados em ensaios bioquímicos.

Nos últimos anos, muitos estudos em diversos patógenos empregaram uma abordagem de genômica subtrativa e relataram identificação e reconhecimento bem-sucedidos de novos alvos terapêuticos específicos para suas respectivas espécies de interesse, tais como Pseudomonas aeruginosa [48], Clostridium botulinum [49], Stenotrophomonas maltophilia [50], Bartonella bacilliformis [51], e Haemophilus ducreyi [52] (Figura 9).

Agora já conhecemos melhor as contribuições que a imunobioinformática pode trazer através da vacinologia reversa e da genômica subtrativa para o combate de agentes infecciosos. Para encerrar, vamos mergulhar em um último exemplo!
Você sabia que a imunobioinformática esteve diretamente envolvida durante a corrida pelas vacinas contra o Covid-19? Não sabia? Pois prepare o álcool em gel, ponha a sua máscara e vamos lá!
Na prática – Sars-CoV-2 e a corrida global por vacinas
A infecção por COVID-19, ou Síndrome Respiratória Aguda Grave por Coronavírus 2 (Sars‐CoV‐2), é uma doença infecciosa causada por um coronavírus recém-descoberto, que já foi responsável por mais de 3,54 milhões de mortes em todo o mundo até maio de 2021 [53].
A OMS declarou uma pandemia em relação ao COVID-19 em 11 de março de 2020, desde então, deixando claro a necessidade urgente de se desenvolver vacinas seguras e eficazes contra essa ameaça global.
Num esforço global para o combate da pandemia, grupos de pesquisa e instituições do mundo todo forneceram serviços, dados e redes de contatos a fim de acelerar o desenvolvimento das vacinas. A plataforma de compartilhamento de dados mais popular contra o COVID-19, chamada GISAID [54], agora hospeda mais de 450.000 genomas virais de SARS-CoV-2, permitindo que métodos in silico de bioinformática pudessem ser aplicados de forma eficaz ao projeto de vacinas, com um tempo de processamento mais rápido do que demandaria a vacinologia clássica.

Nesse caso, duas estratégias imunobioinformáticas principais, vacinologia reversa e vacinologia estrutural (SV), foram aplicadas envolvendo várias etapas (figura 10). Embora RV e SV tenham finalidades diferentes, ambas foram igualmente importantes para desvendar informações sobre os antígenos e a patogênese da doença [55]. Por exemplo, a elucidação estrutural do antígeno de superfície viral trimérico ubíquo (proteína spike) do Sars-CoV-2 (figura 11) foi extremamente importante para os estudos de triagem e modelagem molecular subsequentes e para o desenvolvimento de vacinas de subunidade proteica [56,57].

A produção de vacinas normalmente exige anos de pesquisa e de testes antes de chegar à clínica, mas em 2020, graças a imunobioinformática, os cientistas identificaram em tempo recorde uma vasta gama de candidatos à vacina. Em meados do ano de 2021, 92 vacinas já estavam em ensaios clínicos em humanos, 28 chegaram aos estágios finais de teste. Mundialmente, oito vacinas foram aprovadas para uso e estão sendo disputadas pela maioria dos países [60].
Mas vale lembrar que, enquanto não chega a sua vez de ser vacinado, a melhor maneira de se prevenir e retardar a transmissão da doença ainda é se manter bem-informado. Proteja a si mesmo e as outras pessoas lavando as mãos ou esfregando-as frequentemente com álcool, sem tocar no rosto, e evitando sempre aglomerações.
Conclusão
Apesar dos esforços e avanços da imunobioinformática, vários patógenos persistem sem alternativas viáveis de tratamento. Seja por apresentarem infecções resistentes a múltiplas drogas, por ainda não termos fármacos eficazes contra esses patógenos ou por eles disporem de fatores de virulência que ainda são desconhecidos. Sendo assim, as abordagens de vacinologia reversa e de genômica comparativa, associadas a todas as demais ciências ômicas, são fundamentais para a geração de novas alternativas de prevenção e tratamento, tais como preparações vacinais e alvos farmacológicos.
Portanto, a imunobioinformática se apresenta como grande aliada no combate de doenças vigentes e emergentes, podendo levar à descoberta de novos profiláticos para o tratamento de infecções atuais e a auxiliar na contenção de grandes surtos futuros e epidemias.
Referências
[1] P.J. Delves, S.J. Martin, D.R. Burton, I.M. Roitt, ROITT, FUNDAMENTOS DE IMUNOLOGIA, 2014.
[2] A.K. Abbas, A.H. Lichtman, S. Pillai, Imunologia Celular e Molecular. 8a edição, 2015.
[3] British Society for Immunology, Helper and Cytotoxic T Cells | British Society for Immunology, (2020). https://www.immunology.org/public-information/bitesized-immunology/cells/helper-and-cytotoxic-t-cells (accessed May 31, 2021).
[4] A.C. arl. Goldberg, L.V. icent. Rizzo, MHC structure and function – antigen presentation. Part 1, Einstein (Sao Paulo). 13 (2015) 153–156. https://doi.org/10.1590/S1679-45082015RB3122.
[5] A.R. Santos, V.B. Pereira, E. Barbosa, J. Baumbach, J. Pauling, R. Röttger, M.Z. Turk, A. Silva, A. Miyoshi, V. Azevedo, Mature Epitope Density – A strategy for target selection based on immunoinformatics and exported prokaryotic proteins, BMC Genomics. 14 (2013) S4. https://doi.org/10.1186/1471-2164-14-S6-S4.
[6] J.L. Mellerson, E. Street, C. Knighton, K. Calhoun, R. Seither, J.M. Underwood, Centers for Disease Control and Prevention’s School Vaccination Assessment: Collaboration With US State, Local, and Territorial Immunization Programs, 2012-2018, Am. J. Public Health. 110 (2020) 1092–1097. https://doi.org/10.2105/AJPH.2020.305643.
[7] D.L. Doolan, S.H. Apte, C. Proietti, Genome-based vaccine design: The promise for malaria and other infectious diseases, Int. J. Parasitol. 44 (2014) 901–913. https://doi.org/10.1016/j.ijpara.2014.07.010.
[8] S. Bambini, R. Rappuoli, The use of genomics in microbial vaccine development, Drug Discov. Today. 14 (2009) 252–260. https://doi.org/10.1016/j.drudis.2008.12.007.
[9] Y. He, Z. Xiang, H.L.T. Mobley, Vaxign: The first web-based vaccine design program for reverse vaccinology and applications for vaccine development, J. Biomed. Biotechnol. 2010 (2010) 297505. https://doi.org/10.1155/2010/297505.
[10] E. Del Tordello, R. Rappuoli, I. Delany, Reverse Vaccinology: Exploiting Genomes for Vaccine Design, in: Hum. Vaccines Emerg. Technol. Des. Dev., Elsevier Inc., 2017: pp. 65–86. https://doi.org/10.1016/B978-0-12-802302-0.00002-9.
[11] A. Santos, A. Ali, E. Barbosa, A. Silva, A. Miyoshi, D. Barh, V. Azevedo, The reverse vaccinology – a contextual overview, IIOAB J. 2 (2011) 8–15.
[12] R. Rappuoli, Reverse vaccinology, a genome-based approach to vaccine development, in: Vaccine, Vaccine, 2001: pp. 2688–2691. https://doi.org/10.1016/S0264-410X(00)00554-5.
[13] E. Ong, M.F. Cooke, A. Huffman, Z. Xiang, M.U. Wong, H. Wang, M. Seetharaman, N. Valdez, Y. He, Vaxign2: the second generation of the first Web-based vaccine design program using reverse vaccinology and machine learning, Nucleic Acids Res. (2021). https://doi.org/10.1093/nar/gkab279.
[14] M. Andreatta, M. Nielsen, Gapped sequence alignment using artificial neural networks: Application to the MHC class i system, Bioinformatics. 32 (2016) 511–517. https://doi.org/10.1093/bioinformatics/btv639.
[15] A.P. Bhavsar, J.A. Guttman, B.B. Finlay, Manipulation of host-cell pathways by bacterial pathogens, Nature. 449 (2007) 827–834. https://doi.org/10.1038/nature06247.
[16] M. Sibbald, J.M. van Dijl, Secretome mapping in Gram-positive pathogens, in: 2008: pp. 185-215 BT-Bacterial protein secretion systems.
[17] R. Simeone, D. Bottai, R. Brosch, ESX/type VII secretion systems and their role in host-pathogen interaction, Curr. Opin. Microbiol. 12 (2009) 4–10. https://doi.org/10.1016/j.mib.2008.11.003.
[18] J. Stavrinides, H.C. McCann, D.S. Guttman, Host-pathogen interplay and the evolution of bacterial effectors, Cell. Microbiol. 10 (2008) 285–292. https://doi.org/10.1111/j.1462-5822.2007.01078.x.
[19] J. Couto, G. Seixas, C. Stutzer, N.A. Olivier, C. Maritz-Olivier, S. Antunes, A. Domingos, Probing the rhipicephalus bursa sialomes in potential anti-tick vaccine candidates: A reverse vaccinology approach, Biomedicines. 9 (2021) 363. https://doi.org/10.3390/biomedicines9040363.
[20] M. Pizza, V. Scarlato, V. Masignani, M.M. Giuliani, B. Aricò, M. Comanducci, G.T. Jennings, L. Baldi, E. Bartolini, B. Capecchi, C.L. Galeotti, E. Luzzi, R. Manetti, E. Marchetti, M. Mora, S. Nuti, G. Ratti, L. Santini, S. Savino, M. Scarselli, E. Storni, P. Zuo, M. Broeker, E. Hundt, B. Knapp, E. Blair, T. Mason, H. Tettelin, D.W. Hood, A.C. Jeffries, N.J. Saunders, D.M. Granoff, J.C. Venter, E.R. Moxon, G. Grandi, R. Rappuoli, Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing, Science (80-. ). 287 (2000) 1816–1820. https://doi.org/10.1126/science.287.5459.1816.
[21] S.K. Dhanda, P. Vir, D. Singla, S. Gupta, S. Kumar, G.P.S. Raghava, A Web-Based platform for designing vaccines against existing and emerging strains of mycobacterium tuberculosis, PLoS One. 11 (2016). https://doi.org/10.1371/journal.pone.0153771.
[22] A.S. De Groot, Immunomics: Discovering new targets for vaccines and therapeutics, Drug Discov. Today. 11 (2006) 203–209. https://doi.org/10.1016/S1359-6446(05)03720-7.
[23] F.A. Bidmos, S. Siris, C.A. Gladstone, P.R. Langford, Bacterial vaccine antigen discovery in the reverse vaccinology 2.0 Era: Progress and challenges, Front. Immunol. 9 (2018) 2315. https://doi.org/10.3389/fimmu.2018.02315.
[24] K. Naz, A. Naz, S.T. Ashraf, M. Rizwan, J. Ahmad, J. Baumbach, A. Ali, PanRV: Pangenome-reverse vaccinology approach for identifications of potential vaccine candidates in microbial pangenome, BMC Bioinformatics. 20 (2019) 123. https://doi.org/10.1186/s12859-019-2713-9.
[25] M.A. Ullah, B. Sarkar, S.S. Islam, Exploiting the Reverse Vaccinology Approach to Design Novel Subunit Vaccine against Ebola Virus, Immunobiology. (2020) 2020.01.02.20016311. https://doi.org/10.1101/2020.01.02.20016311.
[26] E.A. Salvador, G.A. Pires de Souza, L.C. Cotta Malaquias, T. Wang, L.F. Leomil Coelho, Identification of relevant regions on structural and nonstructural proteins of Zika virus for vaccine and diagnostic test development: an in silico approach, New Microbes New Infect. 29 (2019) 100506. https://doi.org/10.1016/j.nmni.2019.01.002.
[27] H. Dorosti, M. Eslami, M. Negahdaripour, M.B. Ghoshoon, A. Gholami, R. Heidari, A. Dehshahri, N. Erfani, N. Nezafat, Y. Ghasemi, Vaccinomics approach for developing multi-epitope peptide pneumococcal vaccine, J. Biomol. Struct. Dyn. 37 (2019) 3524–3535. https://doi.org/10.1080/07391102.2018.1519460.
[28] T.C.V. Rodrigues, A.K. Jaiswal, A. De Sarom, L.D.C. Oliveira, C.J.F. Oliveira, P. Ghosh, S. Tiwari, F.M. Miranda, L.D.J. Benevides, V.A.D.C. Azevedo, S.D.C. Soares, Reverse vaccinology and subtractive genomics reveal new therapeutic targets against Mycoplasma pneumoniae: A causative agent of pneumonia, R. Soc. Open Sci. 6 (2019). https://doi.org/10.1098/rsos.190907.
[29] C.L. Araújo, J. Alves, W. Nogueira, L.C. Pereira, A.C. Gomide, R. Ramos, V. Azevedo, A. Silva, A. Folador, Prediction of new vaccine targets in the core genome of Corynebacterium pseudotuberculosis through omics approaches and reverse vaccinology, Gene. 702 (2019) 36–45. https://doi.org/10.1016/j.gene.2019.03.049.
[30] Y. Hisham, Y. Ashhab, Identification of cross-protective potential antigens against pathogenic brucella spp. through combining pan-genome analysis with reverse vaccinology, J. Immunol. Res. 2018 (2018). https://doi.org/10.1155/2018/1474517.
[31] B. Sarkar, M. Ullah, Designing Novel Subunit Vaccines against Herpes Simplex Virus-1 using Reverse Vaccinology Approach, BioRxiv. (2020) 2020.01.10.901678. https://doi.org/10.1101/2020.01.10.901678.
[32] S. Shiragannavar, S. Madagi, J. Hosakeri, V. Barot, In silico vaccine design against Chlamydia trachomatis infection, Netw. Model. Anal. Heal. Informatics Bioinforma. 9 (2020) 39. https://doi.org/10.1007/s13721-020-00243-w.
[33] A.K. Jaiswal, S. Tiwari, S.B. Jamal, D. Barh, V. Azevedo, S.C. Soares, An in silico identification of common putative vaccine candidates against treponema pallidum: A reverse vaccinology and subtractive genomics based approach, Int. J. Mol. Sci. 18 (2017). https://doi.org/10.3390/ijms18020402.
[34] W.H. Organization, World health statistics 2015, World Health Organization, Geneva PP – Geneva, n.d.
[35] P.J. Madabhavi, V.G. Shanmuga Priya, R.N. R, P.S. Honagudi, S. Jiddagi, SUBTRACTIVE GENOMICS – A Promising way To Combat Pathogens (A Review), Int. Res. J. Eng. Technol. (2015) 2395–56.
[36] V. Mountain, Astex, Structural Genomix, and Syrrx, Chem. Biol. 10 (2003) 95–98. https://doi.org/10.1016/S1074-5521(03)00030-9.
[37] T. Hossain, M. Kamruzzaman, T.Z. Choudhury, H.N. Mahmood, A.H.M.N. Nabi, M.I. Hosen, Application of the Subtractive Genomics and Molecular Docking Analysis for the Identification of Novel Putative Drug Targets against Salmonella enterica subsp. enterica serovar Poona, Biomed Res. Int. 2017 (2017). https://doi.org/10.1155/2017/3783714.
[38] D. Barh, S. Tiwari, N. Jain, A. Ali, A.R. Santos, A.N. Misra, V. Azevedo, A. Kumar, In silico subtractive genomics for target identification in human bacterial pathogens, Drug Dev. Res. 72 (2011) 162–177. https://doi.org/10.1002/ddr.20413.
[39] M.I. Hosen, A.M. Tanmoy, D. Al Mahbuba, U. Salma, M. Nazim, M.T. Islam, S. Akhteruzzaman, Application of a subtractive genomics approach for in silico identification and characterization of novel drug targets in Mycobacterium tuberculosis F11, Interdiscip. Sci. Comput. Life Sci. 6 (2014) 48–56. https://doi.org/10.1007/s12539-014-0188-y.
[40] H. Luo, Y. Lin, F. Gao, C.T. Zhang, R. Zhang, DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements, Nucleic Acids Res. 42 (2014). https://doi.org/10.1093/nar/gkt1131.
[41] K.R. Sakharkar, M.K. Sakharkar, V.T.K. Chow, Biocomputational strategies for microbial drug target identification., Methods Mol. Med. 142 (2008) 1–9. https://doi.org/10.1007/978-1-59745-246-5_1.
[42] A.G. Holman, P.J. Davis, J.M. Foster, C.K. Carlow, S. Kumar, Computational prediction of essential genes in an unculturable endosymbiotic bacterium, Wolbachia of Brugia malayi, BMC Microbiol. 9 (2009). https://doi.org/10.1186/1471-2180-9-243.
[43] D. Barh, A.N. Misra, In silico identification of membrane associated candidate drug targets in Neisseria gonorrhoeae, Int. J. Integr. Biol. 6 (2009) 65–67.
[44] T. Sterling, J.J. Irwin, ZINC 15 – Ligand Discovery for Everyone, J. Chem. Inf. Model. 55 (2015) 2324–2337. https://doi.org/10.1021/acs.jcim.5b00559.
[45] M.W. Azam, A. Kumar, A.U. Khan, ACD: Antimicrobial chemotherapeutics database, PLoS One. 15 (2020). https://doi.org/10.1371/journal.pone.0235193.
[46] O. Trott, A.J. Olson, Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem. 31 (2010) 455–461. https://doi.org/10.1002/jcc.21334.
[47] Muniba Faiza, How to perform docking in a specific binding site using AutoDock Vina? | Bioinformatics Review, (2016). https://bioinformaticsreview.com/20161214/how-to-perform-docking-in-a-specific-binding-site-using-autodock-vina/ (accessed June 3, 2021).
[48] R. Uddin, F. Jamil, Prioritization of potential drug targets against P. aeruginosa by core proteomic analysis using computational subtractive genomics and Protein-Protein interaction network, Comput. Biol. Chem. 74 (2018) 115–122. https://doi.org/10.1016/j.compbiolchem.2018.02.017.
[49] R. Sudha, A. Katiyar, P. Katiyar, H. Singh, P. Prasad, Identification of potential drug targets and vaccine candidates in Clostridium botulinum using subtractive genomics approach, Bioinformation. 15 (2019) 18–25. https://doi.org/10.6026/97320630015018.
[50] R.P. Chakrabarty, A.S.M.R.U. Alam, D.K. Shill, A. Rahman, Identification and qualitative characterization of new therapeutic targets in Stenotrophomonas maltophilia through in silico proteome exploration, Microb. Pathog. 149 (2020) 104293. https://doi.org/10.1016/j.micpath.2020.104293.
[51] M.T. Khan, A. Mahmud, A. Iqbal, S.F. Hoque, M. Hasan, Subtractive genomics approach towards the identification of novel therapeutic targets against human Bartonella bacilliformis, Informatics Med. Unlocked. 20 (2020) 100385. https://doi.org/10.1016/j.imu.2020.100385.
[52] A. De Sarom, A.K. Jaiswal, S. Tiwari, L. de C. Oliveira, D. Barh, V. Azevedo, C.J. Oliveira, S. de C. Soares, Putative vaccine candidates and drug targets identified by reverse vaccinology and subtractive genomics approaches to control Haemophilus ducreyi, the causative agent of chancroid, J. R. Soc. Interface. 15 (2018). https://doi.org/10.1098/rsif.2018.0032.
[53] E.O.-O. and J.H. Max Roser, Hannah Ritchie, Coronavirus Pandemic (COVID-19) – the data – Statistics and Research – Our World in Data, OurWorldInData.Org. (2020). https://ourworldindata.org/coronavirus-data (accessed May 31, 2021).
[54] S. Elbe, G. Buckland-Merrett, Data, disease and diplomacy: GISAID’s innovative contribution to global health, Glob. Challenges. 1 (2017) 33–46. https://doi.org/10.1002/gch2.1018.
[55] S. Ishack, S.R. Lipner, Bioinformatics and immunoinformatics to support COVID-19 vaccine development, J. Med. Virol. 1 (2021). https://doi.org/10.1002/jmv.27017.
[56] S. Ismail, S. Ahmad, S.S. Azam, Immunoinformatics characterization of SARS-CoV-2 spike glycoprotein for prioritization of epitope based multivalent peptide vaccine, J. Mol. Liq. 314 (2020) 113612. https://doi.org/10.1016/j.molliq.2020.113612.
[57] J. Ma, D. Su, Y. Sun, X. Huang, Y. Liang, L. Fang, Y. Ma, W. Li, P. Liang, S. Zheng, Cryo-electron Microscopy Structure of S-Trimer, a Subunit Vaccine Candidate for COVID-19, J. Virol. 95 (2021). https://doi.org/10.1128/jvi.00194-21.
[58] RCSB PDB – 7E7D: Cryo-EM structure of the SARS-CoV-2 wild-type S-Trimer from a subunit vaccine candidate, (n.d.). https://www.rcsb.org/structure/7e7d (accessed June 3, 2021).
[59] A.S. Rose, A.R. Bradley, Y. Valasatava, J.M. Duarte, A. Prlic, P.W. Rose, NGL viewer: Web-based molecular graphics for large complexes, Bioinformatics. 34 (2018) 3755–3758. https://doi.org/10.1093/bioinformatics/bty419.
[60] C. Zimmer, Covid-19 Vaccine Tracker: Latest Updates – The New York Times, New York Times. (2021). https://www.nytimes.com/interactive/2020/science/coronavirus-vaccine-tracker.html (accessed May 31, 2021).