
Este artigo tem como objetivo a construção e análise de um interactoma vírus-hospedeiro utilizando dados de interação proteína-proteína. Inicialmente, extraímos da literatura as interações entre as proteínas do SARS-CoV-2 e suas proteínas humanas correspondentes. Posteriormente, essas interações foram enriquecidas com o interactoma humano completo, obtido a partir do banco de dados BIOGRID, adicionando uma camada de complexidade ao modelo. Após a construção do interactoma viral, por meio de um procedimento detalhado no software Cytoscape, realizamos uma análise de enriquecimento funcional das proteínas humanas que interagem com a proteína S do vírus, utilizando a ferramenta Enrichr e a base de dados WikiPathways 2024. Esse processo visa identificar as principais vias biológicas envolvidas na infecção viral. Além disso, exploramos análises adicionais que podem ser realizadas a partir do interactoma completo, como a identificação de genes hub, os quais desempenham papéis cruciais no controle da rede de interações. A integração entre o interactoma específico do vírus e o interactoma humano completo proporciona uma visão mais ampla das consequências da infecção viral nas redes proteicas humanas, oferecendo insights fundamentais para o desenvolvimento de estratégias terapêuticas mais eficazes.
Autor: Lucas Miguel de Carvalho (ORCID: 0000-0002-8766-0452)
Revisão: Aline de Paula Dias da Silva [ORCID: 0009-0003-5022-0642], Isaac, Carlos Capelini
1. Interactoma vírus-hospedeiro
O estudo das interações entre vírus e hospedeiro utilizando interactomas desempenha um papel fundamental na compreensão dos mecanismos subjacentes às infecções virais. Ao contrário dos organismos celulares, os vírus apresentam uma enorme diversidade em termos de composição genômica, evolução e funções proteicas. Essa variação influencia diretamente como os vírus interagem com as proteínas do hospedeiro, levando a adaptações específicas para garantir sua replicação e sobrevivência [1]. As interações entre proteínas virais e do hospedeiro são frequentemente mediadas por interações entre domínios protéicos, onde os vírus, muitas vezes, utilizam interfaces exógenas para competir com as interações endógenas do hospedeiro, desencadeando uma corrida evolutiva entre ambos os sistemas [2].
As interações entre proteínas (protein-protein interactions – PPIs) são frequentemente modeladas e analisadas utilizando a teoria dos grafos, na qual as proteínas ou genes são representados como nós e as interações entre eles como arestas. Esse formalismo permite a construção de redes de interação proteica que oferecem uma visão global dos processos celulares, facilitando a compreensão dos mecanismos biológicos em nível sistêmico. Estudos demonstram que proteínas específicas podem desempenhar papéis cruciais nessas redes, funcionando como alvos potenciais para intervenções terapêuticas [3,4]. Dessa forma, redes de interação proteica são componentes essenciais tanto para a modelagem de processos celulares quanto para a descoberta de novos fármacos.
Ao mapear essas interações em redes complexas de proteínas, conhecidas como interactomas, podemos identificar os nós centrais (hubs) e “gargalos” (bottlenecks), que são frequentemente os alvos de patógenos. Os vírus tendem a explorar esses nós altamente conectados para desestabilizar o interactoma do hospedeiro, influenciando processos fundamentais e facilitando a infecção, replicação e evasão imunológica. Além disso, a análise das propriedades topológicas dessas redes, como a conectividade e centralidade, revela como os vírus reestruturam as interações moleculares para maximizar sua eficiência patogênica. Estudos desses interactomas não só ampliam nossa compreensão dos mecanismos virais, mas também oferecem insights cruciais para o desenvolvimento de estratégias terapêuticas e preparam a ciência para responder a futuras pandemias.
Dessa forma, o uso de interactomas para estudar as interações vírus-hospedeiro é essencial para identificar novas oportunidades de intervenção médica, bem como para elucidar os padrões de interação que sustentam a patogênese viral.
2. Dados de entrada
Para o nosso exemplo inicial de interação vírus-hospedeiro, utilizaremos os dados do interactoma vírus-hospedeiro fornecidos por Das, JK e colaboradores (2021) no estudo “Analyzing host-viral interactome of SARS-CoV-2 for identifying vulnerable host proteins during COVID-19 pathogenesis“, publicado na revista Infect Genet Evol. Esse estudo disponibiliza um conjunto de dados que detalha as interações entre o vírus SARS-CoV-2 e o sistema humano. A escolha deste banco de dados se deve à sua relevância e abrangência nas interações do vírus com proteínas humanas, que são fundamentais para a compreensão dos mecanismos de patogênese do COVID-19.
Além disso, ao integrar os dados do interactoma vírus-hospedeiro com o interactoma completo humano, poderemos identificar interações adicionais que vão além das que conectam diretamente às proteínas do vírus. Isso permitirá explorar interações indiretas ou potenciais novos alvos terapêuticos que não foram capturados pelo estudo original. Enquanto o estudo de Das et al. foca especificamente nas interações entre o SARS-CoV-2 e proteínas humanas diretamente envolvidas no ciclo viral, o estudo proposto neste artigo amplia essa análise, considerando um espectro mais amplo de interações, o que pode fornecer uma visão mais holística do impacto do vírus no organismo hospedeiro.
Também faremos o download do interactoma humano completo a partir do banco de dados BIOGRID [5]. Esse conjunto de dados será utilizado para reconstruir a rede completa de interações proteicas no contexto humano, permitindo uma análise mais ampla e detalhada das interações mediadas pelo SARS-CoV-2.
Este artigo tem como propósito construir um interactoma vírus-humano utilizando bancos de dados de interações proteína-proteína, se baseando no pipeline mostrado na Figura 1. As análises de redes serão realizadas no Cytoscape v3.10.2 [6].
 Figura 1. Elaboração do interactoma humano-viral. Esta figura ilustra o pipeline de análise utilizado para construir o interactoma humano-viral. Inicialmente, temos em mãos os dados do interactoma vírus-hospedeiro. A partir desse ponto, é realizada uma integração com o interactoma completo humano, que inclui todas as proteínas humanas disponíveis em bancos de dados públicos. O resultado final do pipeline é um conjunto expandido de interações que pode fornecer novos insights sobre os mecanismos de patogênese e potenciais alvos terapêuticos.
Figura 1. Elaboração do interactoma humano-viral. Esta figura ilustra o pipeline de análise utilizado para construir o interactoma humano-viral. Inicialmente, temos em mãos os dados do interactoma vírus-hospedeiro. A partir desse ponto, é realizada uma integração com o interactoma completo humano, que inclui todas as proteínas humanas disponíveis em bancos de dados públicos. O resultado final do pipeline é um conjunto expandido de interações que pode fornecer novos insights sobre os mecanismos de patogênese e potenciais alvos terapêuticos.
3. Gerando o interactoma vírus-humano
No Material Suplementar A disponibilizado por Das et al. 2021 [7], encontramos o interactoma vírus-hospedeiro para o SARS-CoV-2, contendo informações valiosas sobre as interações entre as proteínas virais e humanas. É importante destacar que o pipeline descrito a seguir pode ser adaptado para qualquer tipo de interactoma vírus-hospedeiro, oferecendo flexibilidade para diferentes estudos.
O primeiro passo consiste em mapear as interações entre as proteínas do vírus e do hospedeiro humano. Por exemplo, neste dataset, encontramos a conhecida interação entre a proteína S (Spike) do SARS-CoV-2 e a proteína ECA2 humana, essencial para a entrada do vírus nas células. No total, o interactoma vírus-hospedeiro inclui 2489 interações.
Além disso, para complementar a análise, baixaremos o interactoma humano completo a partir do banco de dados BIOGRID. Para acessar esses dados, basta seguir o caminho: Downloads >> Current-release >> BIOGRID-ORGANISM-4.4.238.tab.zip. Após extrair o arquivo, localize a tabela intitulada BIOGRID-ORGANISM-Homo_sapiens-4.4.238.tab, que contém as interações proteicas. Focaremos nas duas primeiras colunas da tabela, OFFICIAL_SYMBOL_A e OFFICIAL_SYMBOL_B, que representam os IDs das proteínas que interagem entre si. O dataset completo do BIOGRID para Homo sapiens conta com 1.248.887 interações, fornecendo um rico conjunto de dados para a reconstrução do interactoma humano.
Com esses dois conjuntos de dados, seremos capazes de realizar uma análise integrada das interações vírus-hospedeiro, permitindo uma compreensão mais profunda da dinâmica molecular durante a infecção.
Passo 1: Carregando ambos interactomas no Cytoscape
Dificuldade: média
Para importar a rede do artigo de Das, et al. 2021, basta ir no Cytoscape em File >> Import >> Network from file, selecionar o arquivo, e selecionar a coluna e seu tipo (source e target). Quando carregar a rede (Figura 2-A), teremos duas colunas “shared name” e “name”. A primeira será utilizada para a junção das redes. A Figura 2-B mostra um zoom da tela do Cytoscape apresentando a interação entre os genes nps15 e SPIRE1.
Ao carregar o interactoma humano do BIOGRID, muito cuidado se você tem menos de 8Gb de RAM no seu computador, pois ele irá travar, certamente. A ideia é a mesma da anterior, bastando ir em File >> Import >> Network from file, e selecionando as colunas OFFICIAL_SYMBOL_A e OFFICIAL_SYMBOL_B como target e source, respectivamente. Um conselho é: não crie o view do interactoma, pois, caso contrário, creio que a tela azul será o próximo passo.
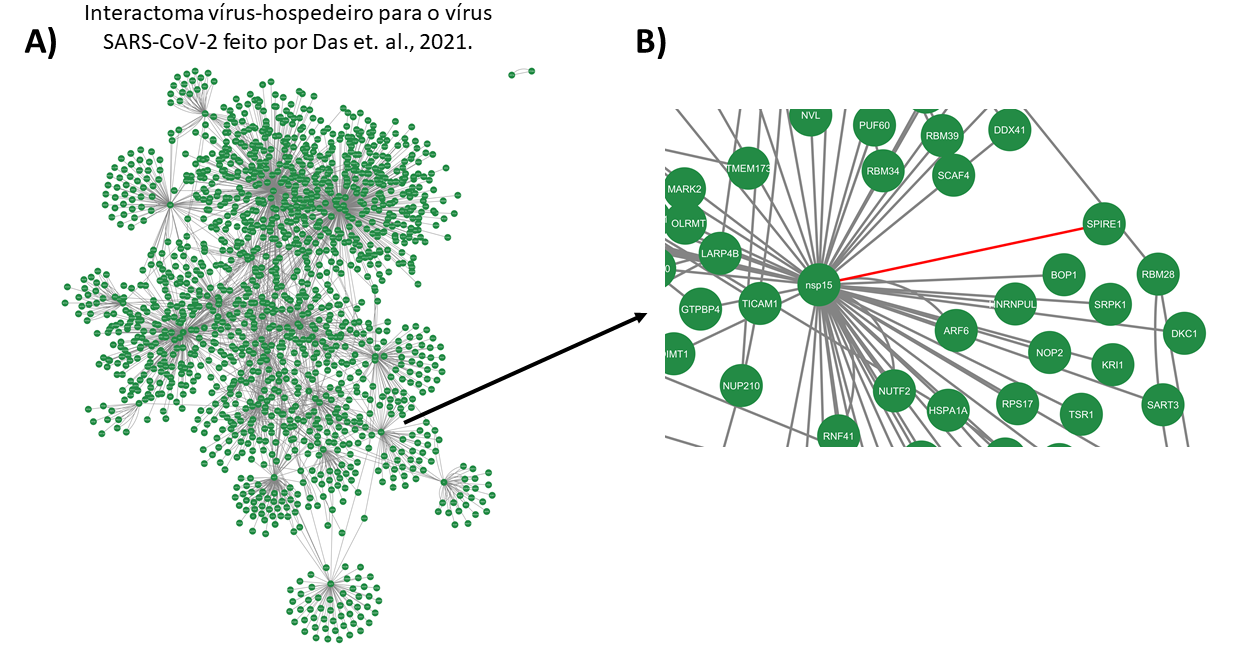 Figura 2. Importação e visualização da rede de interação vírus-hospedeiro no Cytoscape. (A) Interactoma vírus-hospedeiro para o vírus SARS-CoV-2 elaborado por Das, et al., 2021. (B) Exemplo de visualização da interação entre os genes nps15 e SPIRE1. A imagem mostra um zoom na tela do Cytoscape, destacando a interação específica entre essas duas proteínas, conforme identificado na rede importada.
Figura 2. Importação e visualização da rede de interação vírus-hospedeiro no Cytoscape. (A) Interactoma vírus-hospedeiro para o vírus SARS-CoV-2 elaborado por Das, et al., 2021. (B) Exemplo de visualização da interação entre os genes nps15 e SPIRE1. A imagem mostra um zoom na tela do Cytoscape, destacando a interação específica entre essas duas proteínas, conforme identificado na rede importada.
Passo 2: Criando o interactoma humano-viral
Dificuldade: média
Neste momento, temos os dois interactomas carregados no Cytoscape, como mostra o início da Figura 1. Para realizar o passo de juntar ambos (MERGE), vamos executar no Cytoscape o comando Tools >> Merge >> Networks. Neste ponto devemos selecionar ambas as colunas que devem ser juntadas e, como dito anteriormente, vamos usar a coluna “shared name” de ambas. O tipo de fusão foi designado “Union”, pois o objetivo deste projeto era integrar os genes do SARS-CoV-2 com os do genoma humano. Muito cuidado que este passo novamente requer um grande processamento de memória.
Após montar o interactoma vírus-hospedeiro final para o SARS-CoV-2, podemos verificar como estão as interações com as proteínas do vírus, como a proteína S (Figura 3), e que estão presentes no interactoma humano. É importante neste momento, além de analisar as proteínas humanas que estão diretamente ligadas às proteínas virais, mas também aquelas que são agregadas a estas proteínas, e as interacções entre as proteínas do interactoma vírus-hospedeiro original que não existiam.
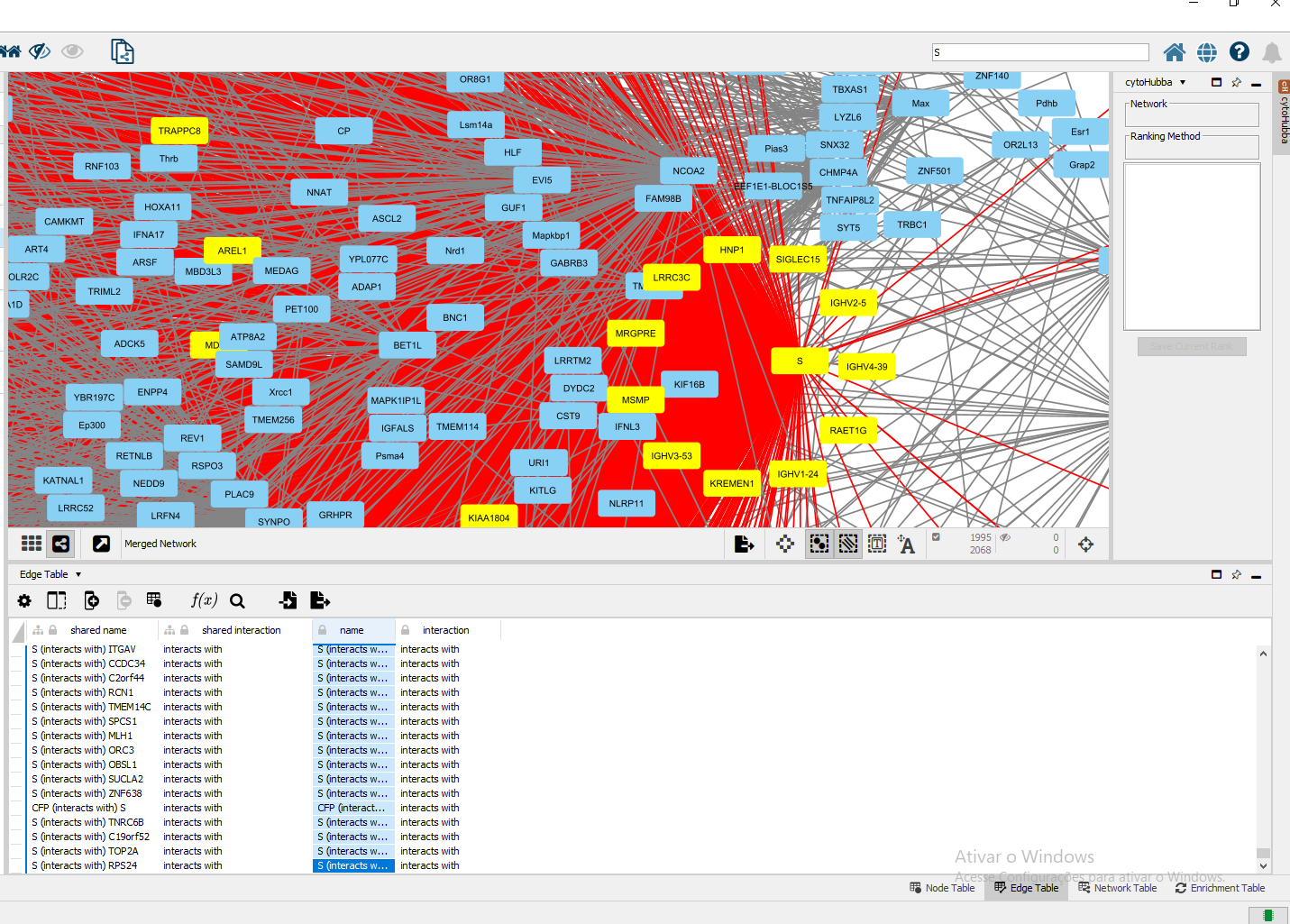 Figura 3. Destaque para a proteína S do vírus SARS-CoV-2 dentro do interactoma vírus-hospedeiro com humano.
Figura 3. Destaque para a proteína S do vírus SARS-CoV-2 dentro do interactoma vírus-hospedeiro com humano.
Passo 3. Extraindo uma sub rede do interactoma
Dificuldade: média
As proteínas virais do SARS-CoV-2, como a proteína S, foram pesquisadas utilizando a ferramenta de busca na rede, e seus respectivos nós foram afastados do interactoma vírus-hospedeiro principal. Isso garante uma identificação mais fácil do gene na criação de redes subsequentes.
Para o nosso exemplo sobre o SARS-CoV-2, a principal proteína S foi selecionada, junto com seus interatores diretos (primeiros vizinhos). Isso foi feito buscando essa proteína S dentro da rede e selecionando seus primeiros vizinhos. Uma vez que todos os nós de interesse foram selecionados, uma nova rede é criada selecionando File > New > Network > from Selected nodes, Selected edges. Isso produz uma rede secundária menor, como mostrado na Figura 4. Esse interactoma contém os genes que interagem diretamente com a proteína S e sua relação com outros genes humanos. A rede possui 1995 nós. Isso mostra que o interactoma do BIOGRID introduziu mais interações do que inicialmente estava descrito por Das, et al. 2021, além das próprias interações entre as proteínas que são primeiras vizinhas da proteína S. Isso é importante, pois quaisquer métricas que forem calculadas sobre a rede, as interações entre as proteínas humanas, além das interações entre proteínas virais e humanas, devem ser levadas em conta, e traz maior sentido biológico à rede.
Destacamos que a aplicação deste pipeline se estende a integração de ômicas quando, por exemplo, queremos adicionar informações à rede metabólito-proteína. Também se estende na seleção de proteínas de interesse, sendo até comum selecionar mais de uma proteína viral para montar uma sub rede.
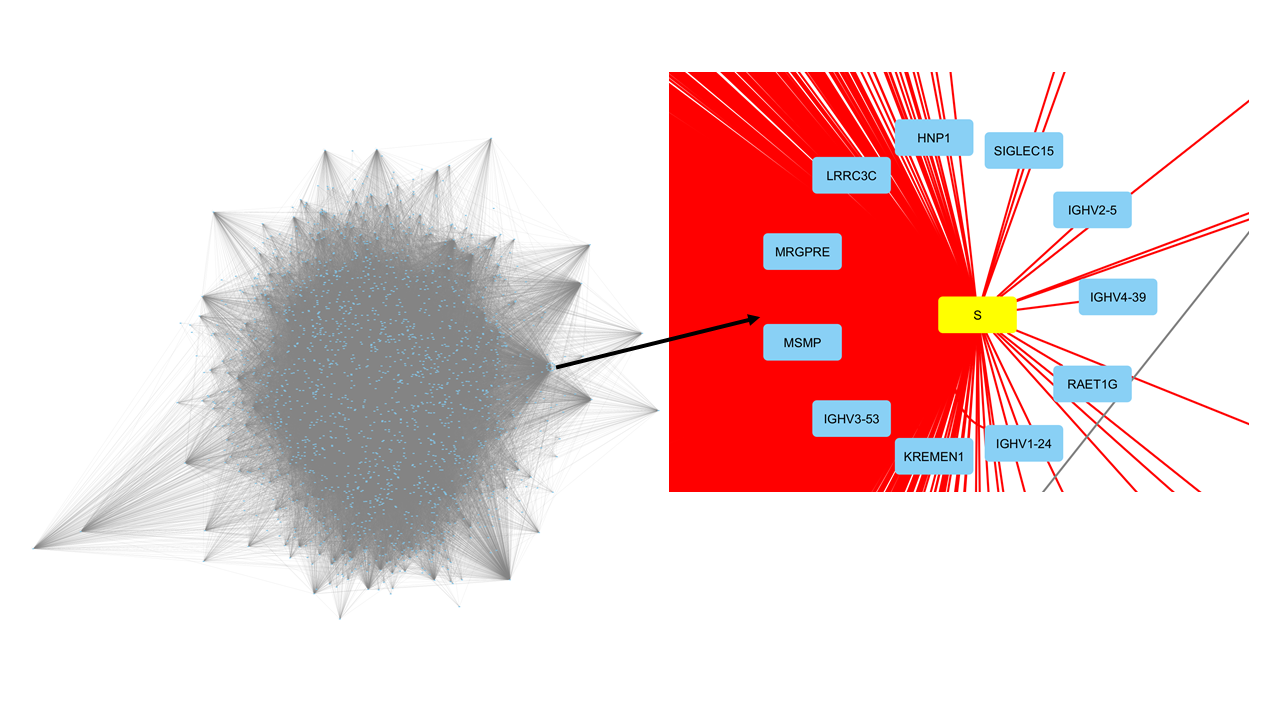 Figura 4. Interactoma extraído sobre as proteínas que interagem com a proteína S do vírus SARs-CoV-2, destacada em amarelo.
Figura 4. Interactoma extraído sobre as proteínas que interagem com a proteína S do vírus SARs-CoV-2, destacada em amarelo.
Passo 4. Enriquecimento biológico da sub rede
Dificuldade: média
Extraímos as proteínas humanas que interagem diretamente com a proteína S (Spike) do SARS-CoV-2, a partir do interactoma vírus-hospedeiro final. Essas proteínas foram utilizadas para realizar uma análise de enriquecimento funcional por meio da ferramenta Enrichr [8], utilizando a biblioteca WikiPathways 2024. Esse tipo de análise nos permite identificar quais vias biológicas estão mais associadas às proteínas que interagem com a proteína S, proporcionando uma visão mais abrangente dos processos celulares potencialmente afetados pelo vírus. No total foram identificadas 193 vias enriquecidas (p-adj < 0.05), onde, dentre elas, selecionamos algumas vias interessantes, como mostra a Tabela 1.
Tabela 1. Resultado do enriquecimento por WikiPathways dos genes que interagem com a proteína S (Spike) do SARS-CoV-2 no interactoma vírus-hospedeiro final.
|
Termo |
Razão |
P-valor ajustado |
|
Network Map Of SARS CoV 2 Signaling WP5115 |
67/254 |
1.38E-11 |
|
Sterol Regulatory Element Binding Proteins SREBP Signaling WP1982 |
26/69 |
9.41E-08 |
|
Arrhythmogenic Right Ventricular Cardiomyopathy WP2118 |
27/74 |
9.41E-08 |
|
Cholesterol Metabolism WP5304 |
26/72 |
2.18E-07 |
|
Brain Derived Neurotrophic Factor BDNF Signaling WP2380 |
34/137 |
1.27E-05 |
|
TCA Cycle And Deficiency Of Pyruvate Dehydrogenase Complex PDHc WP2453 |
10/16 |
1.27E-05 |
|
TGF Beta Signaling Pathway WP366 |
26/132 |
0.004698707 |
|
Axon Guidance WP5289 |
17/72 |
0.004966206 |
|
IL10 Anti Inflammatory Signaling WP4495 |
05/12 |
0.023307764 |
|
Resistin As A Regulator Of Inflammation WP4481 |
09/33 |
0.022180242 |
A partir da rede gerada, é possível realizar análises mais detalhadas para identificar genes hub, que desempenham um papel crucial na regulação e controle da rede de interações. Essas análises são fundamentais para destacar, além da proteína S, outros genes e proteínas humanas que possam ter relevância no processo de infecção viral e na resposta do hospedeiro. Dessa forma, genes identificados como hubs podem ser investigados como potenciais alvos terapêuticos ou biomarcadores para o desenvolvimento de tratamentos antivirais.
4. Conclusão
A integração do interactoma vírus-hospedeiro com o interactoma humano completo representa um passo crucial para aprofundar nossa compreensão sobre os mecanismos moleculares subjacentes à infecção viral. Enquanto o interactoma vírus-hospedeiro nos fornece informações diretas sobre as interações entre as proteínas virais e suas correspondentes proteínas humanas, a adição do interactoma humano completo permite expandir essas conexões, revelando interações secundárias e redes mais amplas que podem ser influenciadas pela infecção. Essa abordagem agrega complexidade ao modelo, permitindo uma análise mais detalhada de como a infecção viral pode repercutir nas redes proteicas humanas e identificar possíveis alvos terapêuticos adicionais. Assim, essa integração pode auxiliar no desenvolvimento de estratégias mais eficazes de intervenção, melhorando nossa capacidade de responder a pandemias e infecções virais.
5. Referências
1. Halehalli, R.R.; Nagarajaram, H.A. Molecular Principles of Human Virus Protein-Protein Interactions. Bioinformatics 2015, 31, 1025–1033, doi:10.1093/BIOINFORMATICS/BTU763.
2. Brito, A.F.; Pinney, J.W. Protein-Protein Interactions in Virus-Host Systems. Front Microbiol 2017, 8, doi:10.3389/FMICB.2017.01557/FULL.
3. Li, W.; Chen, L.; He, W.; Li, W.; Qu, X.; Liang, B.; Gao, Q.; Feng, C.; Jia, X.; Lv, Y.; et al. Prioritizing Disease Candidate Proteins in Cardiomyopathy-Specific Protein-Protein Interaction Networks Based on “Guilt by Association” Analysis. PLoS One 2013, 8, doi:10.1371/JOURNAL.PONE.0071191.
4. Ferrari, R.; Kia, D.A.; Tomkins, J.E.; Hardy, J.; Wood, N.W.; Lovering, R.C.; Lewis, P.A.; Manzoni, C. Stratification of Candidate Genes for Parkinson’s Disease Using Weighted Protein-Protein Interaction Network Analysis. BMC Genomics 2018, 19, doi:10.1186/S12864-018-4804-9/METRICS.
5. Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID Database: A Comprehensive Biomedical Resource of Curated Protein, Genetic, and Chemical Interactions. Protein Science 2021, 30, 187–200, doi:10.1002/PRO.3978.
6. Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res 2003, 13, 2498, doi:10.1101/GR.1239303.
7. Das, J.K.; Roy, S.; Guzzi, P.H. Analyzing Host-Viral Interactome of SARS-CoV-2 for Identifying Vulnerable Host Proteins during COVID-19 Pathogenesis. Infection, Genetics and Evolution 2021, 93, 104921, doi:10.1016/J.MEEGID.2021.104921.
8. Kuleshov, M. V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res 2016, 44, W90–W97, doi:10.1093/NAR/GKW377.