
Este artigo apresenta um tutorial de uso da ferramenta PatternLab for Proteomics V. Boa leitura!
Autores: Lucas Marques da Cunha, Rafaela Marie Melo da Cunha, Giullia de Souza Santos
Revisão: Bruna Espino, Tati Bialves, Luana bastos
INTRODUÇÃO
As proteínas são responsáveis por diversas funções estruturais, regulatórias e catalíticas nas células. O conjunto completo dessas proteínas, incluindo suas interações e variações, compõem o proteoma, uma representação dinâmica e complexa da atividade molecular celular. A espectrometria de massas (MS) permite a identificação e quantificação de milhares de proteínas em diferentes amostras e contextos experimentais. Essa tecnologia é crucial para o avanço na compreensão dos processos biológicos e no estudo de várias doenças (SINITCYN; RUDOLPH; COX, 2018).
A proteômica baseada em espectrometria de massas pode ser dividida em duas principais áreas: a identificação e quantificação de peptídeos e proteínas, e a análise downstream, voltada para a interpretação biológica dos dados gerados (GAO; YATES, 2019). No entanto, a detecção de peptídeos variantes ainda enfrenta desafios significativos. Embora o sequenciamento de nova geração tenha levado à descoberta de milhares de mutações associadas ao câncer, a análise proteômica de isoformas permanece limitada. Apenas uma pequena fração das variantes de nucleotídeo único identificadas por sequenciamento de DNA e RNA é confirmada no nível proteico. Isso se deve a barreiras técnicas que dificultam a detecção precisa dessas alterações, prejudicando a integração completa entre os dados genômicos e proteômicos (LIN et al., 2023). Além disso, o splicing alternativo contribui significativamente para essa dificuldade, pois gera isoformas proteicas distintas que podem mascarar ou impedir a identificação de variantes no nível proteico, complicando ainda mais a análise e correlação entre as informações genéticas e proteicas (FESENKO et al., 2017).
Um dos maiores obstáculos na identificação de variantes proteicas em experimentos de proteômica shotgun é a dependência de buscas em bancos de dados de sequências proteicas. Muitos desses bancos não incluem informações sobre variantes de proteínas, o que impede a identificação de peptídeos e proteínas mutantes. A inclusão de variações de codificação conhecidas nesses bancos pode ajudar a superar esse desafio. Como resposta, foram criados recursos como o Banco de Dados de Variação do Proteoma do Câncer, que anota de maneira abrangente milhares de variantes relacionadas ao câncer, além de variantes não específicas da doença, coletadas no Banco de Dados de Polimorfismo de Nucleotídeo Único (dbSNP). Esses bancos de dados são fundamentais para melhorar a detecção de peptídeos variantes, acelerando o desenvolvimento de biomarcadores e novos alvos terapêuticos (LI et al., 2011).
Na proteômica bottom-up, ou shotgun, a análise começa pela digestão das proteínas em peptídeos, o que permite uma análise aprofundada do proteoma (DEN RIDDER; DARAN-LAPUJADE; PABST, 2020). Este método é amplamente utilizado, especialmente em pesquisas que investigam alterações proteicas em doenças complexas, como o câncer.
Entre as ferramentas computacionais disponíveis para a análise de dados proteômicos, destaca-se o PatternLab for Proteomics V, que integra diversos módulos em um ambiente independente, facilitando desde a correspondência de espectros até análises estatísticas avançadas e visualização de dados (CARVALHO et al., 2008; SANTOS et al., 2022). O PatternLab foi desenvolvido para tornar a análise de dados proteômicos mais acessível, com uma interface gráfica amigável e altamente funcional (CARVALHO et al., 2008; SANTOS et al., 2022). Este tutorial oferece um guia abrangente sobre o uso do PatternLab V, com ênfase em sua aplicação na pesquisa do câncer, demonstrando como o software pode enfrentar desafios importantes, como a identificação de peptídeos variantes e a organização eficiente de grandes volumes de dados.
PATTERNLAB FOR PROTEOMICS V: UMA FERRAMENTA PARA PROTEÔMICA SHOTGUN
O PatternLab for Proteomics V é um ambiente computacional integrado, gratuito e multiplataforma, desenvolvido para simplificar a análise de dados proteômicos shotgun (CARVALHO et al., 2008). Ele permite a execução de diversas tarefas, como formatação de bancos de dados de sequências, análise de espectros de peptídeos, filtragem estatística e visualização gráfica dos resultados. Essa ferramenta foi desenvolvida pelo Laboratório de Proteômica Computacional e Estrutural – Fiocruz (https://www.icc.fiocruz.br/lpec/o-laboratorio/), coordenado pelos docentes Paulo Carvalho e Tatiana Brasil. Abaixo, apresentamos um guia passo a passo para instalação e configuração inicial.
Passo 1 – Download do Software: O PatternLab for Proteomics V pode ser baixado gratuitamente no site oficial: http://patternlabforproteomics.org. O software está disponível exclusivamente para a plataforma Windows. Além disso, é necessário instalar o Visual C++ Runtime 14 para que o PatternLab funcione corretamente. O pacote de instalação do Visual C++ pode ser baixado diretamente na página de suporte da Microsoft: https://aka.ms/vs/16/release/vc_redist.x64.exe. A página inicial do site fornece orientações detalhadas para o download, bem como informações sobre ferramentas auxiliares necessárias para o pleno funcionamento do software (ver Figura 1).
 Figura 1 – Página Inicial do Pattern Lab for Proteomics V com link para download da ferramenta.
Figura 1 – Página Inicial do Pattern Lab for Proteomics V com link para download da ferramenta.
Passo 2 – Requisitos de Sistema: antes de iniciar a instalação, é importante verificar se seu sistema atende aos seguintes requisitos mínimos:
-
Sistema Operacional: Windows 10 ou superior.
-
Processador: Intel Core i5 ou superior.
-
Memória RAM: 8 GB (16 GB recomendados para grandes conjuntos de dados).
-
Espaço em Disco: 2 GB para instalação básica.
-
Conexão com a Internet: Necessária para baixar atualizações e módulos adicionais.
Passo 3 – Processo de Instalação: execute o arquivo .exe que foi baixado e siga as instruções do assistente de instalação. Caso seja necessário, o sistema solicitará a instalação de componentes adicionais (Figura 2-A). Após essa confirmação, o processo de instalação da ferramenta começará, realizando o download dos módulos (Figura 2-B). Após a conclusão, o software será iniciado automaticamente. Na página inicial, serão exibidas as atualizações dos módulos e correções de erros recentes.
 Figura 2 – Processo de Instalação do Pattern Lab for Proteomics. A) Instalação de componentes adicionais. B) Download e instalação dos módulos da ferramenta.
Figura 2 – Processo de Instalação do Pattern Lab for Proteomics. A) Instalação de componentes adicionais. B) Download e instalação dos módulos da ferramenta.
A Figura 3 apresenta uma visão geral da ferramenta após a instalação. No menu “Load”, é possível carregar resultados de buscas anteriores que foram salvos, além de importar e visualizar os dados de análises provenientes de plataformas de espectrometria de massas. O exemplo ilustrado na Figura 4 apresenta dados extraídos do banco de dados Proteomic Data Commons (PDC): https://proteomic.datacommons.cancer.gov/pdc/ (THANGUDU et al., 2022), com ID=PDC000111 e SAMPLE=TCGA-A6-3808-01A, referente a um caso de câncer colorretal. Na parte superior, é exibido o resultado da cromatografia ao longo do tempo, com o eixo x representando o tempo e o eixo y a intensidade relativa. Ao clicar em qualquer ponto da cromatografia, o espectro de massa correspondente é exibido na parte inferior da figura. Além disso, é possível ampliar os espectros clicando diretamente sobre o espectro desejado para uma visualização mais detalhada.  Figura 3 – Visão geral da ferramenta Pattern Lab for proteomics.
Figura 3 – Visão geral da ferramenta Pattern Lab for proteomics.
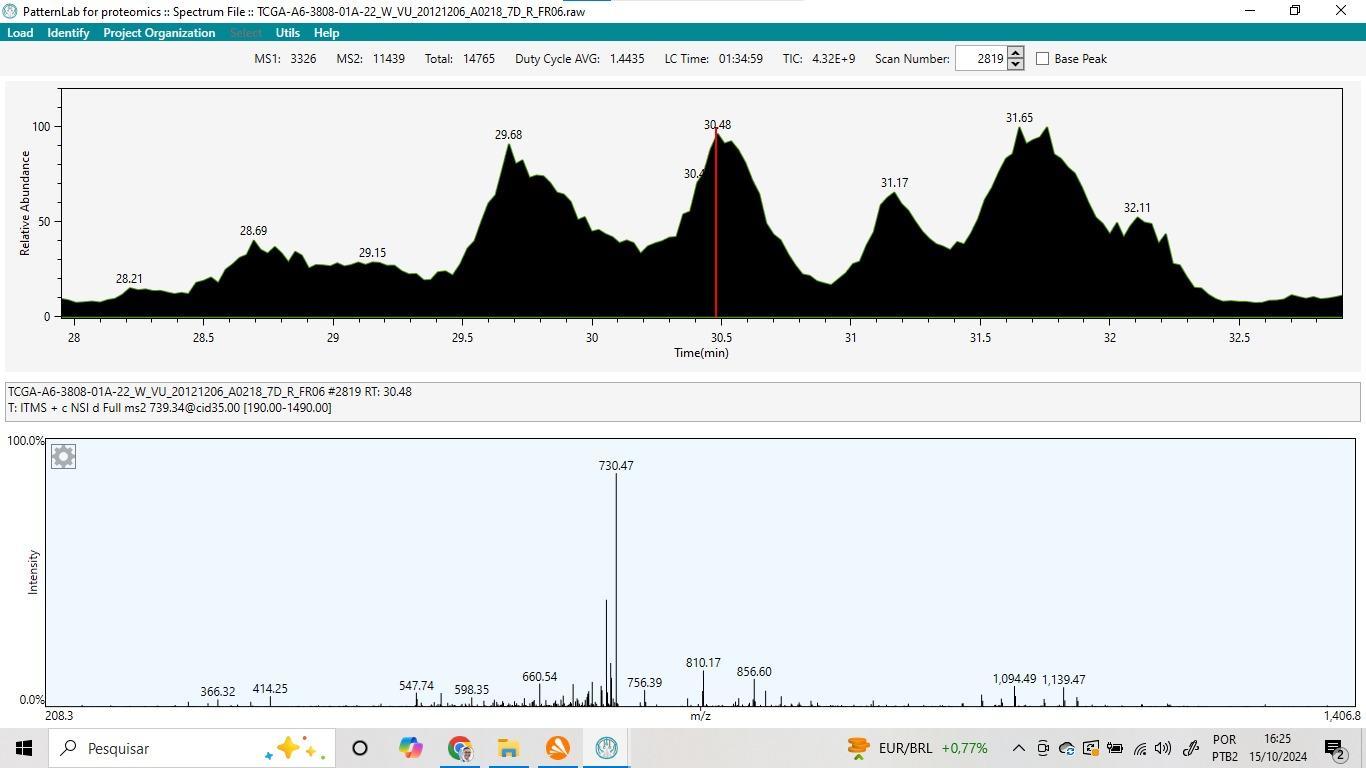 Figura 4 – Visualização do resultado do experimento de espectrometria de massas provenientes do PDC.
Figura 4 – Visualização do resultado do experimento de espectrometria de massas provenientes do PDC.
O menu “Identify” é o local onde é possível identificar proteínas por meio de correspondência entre os espectros experimentais e os espectros teóricos. Para isso, basta acessar a opção “Peptide Spectrum Matching Search and Filtering” e definir os parâmetros de busca tais como equipamento utilizado, modificações químicas, diretório contendo os espectros experimentais e o arquivo fasta contendo o banco de dados de proteínas (Ver Figura 5).
A ferramenta também permite selecionar a opção YADA3 (Yet Another Deconvolution Algorithm 3.0) que é utilizada na espectrometria de massas para realizar a deconvolução de espectros de alta resolução. Ele auxilia na análise de dados complexos, separando picos isotópicos e determinando os estados de carga e massas monoisotópicas (CARVALHO et al., 2008). Isso permite identificar com precisão proteínas e peptídeos, sendo especialmente útil na proteômica, onde se trabalha com moléculas grandes e dados complexos (CARVALHO et al., 2008).
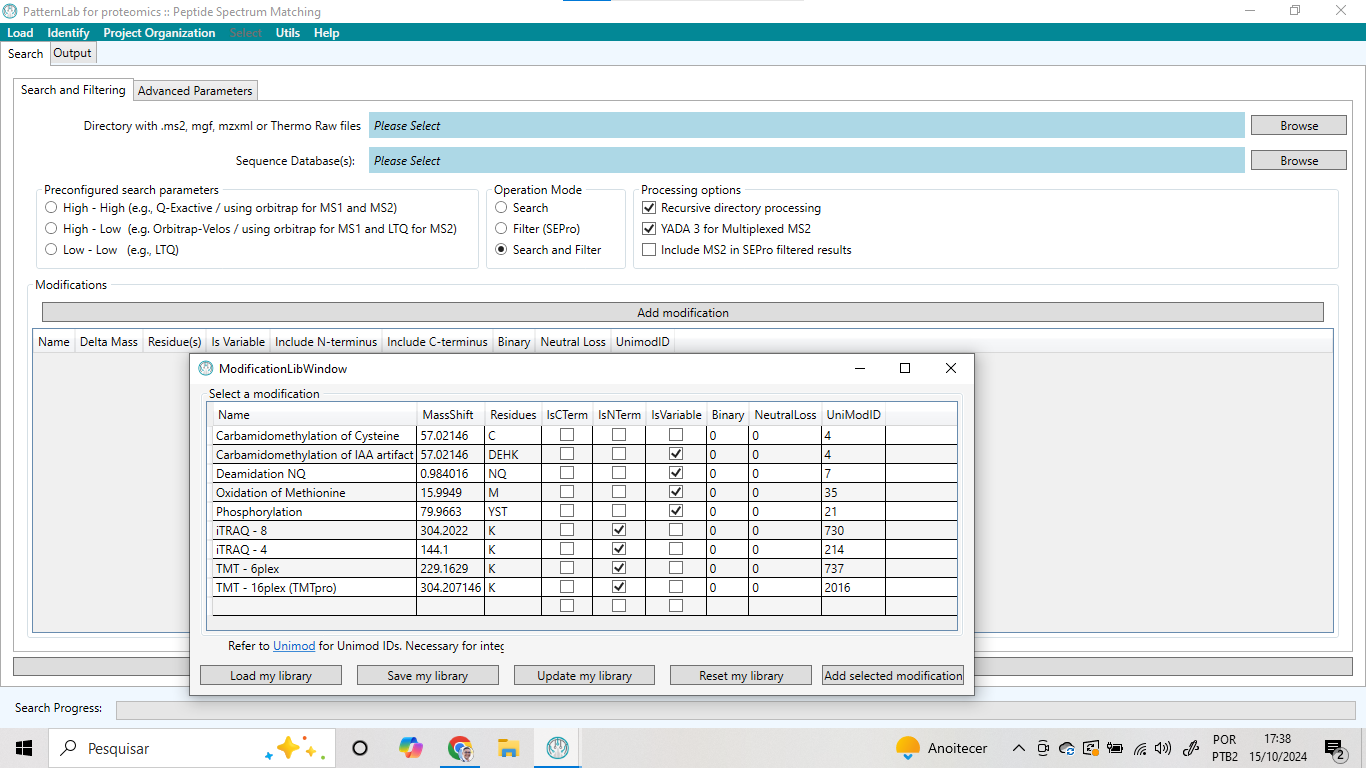 Figura 5 – Visão geral da opção “Peptide Spectrum Matching Search and Filtering” presente no menu “Identify”.
Figura 5 – Visão geral da opção “Peptide Spectrum Matching Search and Filtering” presente no menu “Identify”.
A configuração de parâmetros avançados no PatternLab for Proteomics e seu motor de busca Comet permite ajustar a análise de espectros MS/MS de forma precisa (Ver Figura 6). Ao compreender como cada um desses parâmetros afeta os resultados, você pode otimizar suas buscas e obter identificações de peptídeos e proteínas mais precisas. Dependendo do experimento, ajustes adicionais podem ser necessários, especialmente em relação à tolerância de massa, enzima e modificações variáveis.
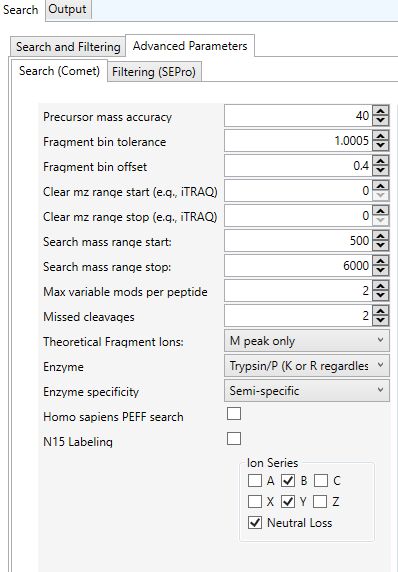 Figura 6 – Parâmetros avançados da ferramenta Comet presentes na opção “Peptide Spectrum Matching Search and Filtering” do menu “Identify”.
Figura 6 – Parâmetros avançados da ferramenta Comet presentes na opção “Peptide Spectrum Matching Search and Filtering” do menu “Identify”.
Abaixo, será apresentada uma explicação de cada parâmetro com foco em suas funcionalidades e exemplos práticos de uso.
-
Precursor mass accuracy (Precisão da massa do precursor): Descrição: Define a tolerância de massa para os íons precursores (peptídeos antes da fragmentação). O valor está definido como 40 ppm (partes por milhão). Isso significa que o software aceita uma diferença de até 40 ppm entre a massa calculada do peptídeo e a massa medida experimentalmente. É recomendado usar valores menores (como 10 ou 20 ppm) para dados de alta resolução (espectrômetros de massa como Orbitrap) para aumentar a precisão da busca (CARVALHO et al., 2008; CARVALHO et al., 2009; ENG; JAHAN; HOOPMANN, 2013;SANTOS et al., 2022; CLASEN et al., 2023).
-
Fragment bin tolerance (Tolerância da massa dos fragmentos): define a janela de tolerância para os íons fragmentos no espectro MS/MS. O valor padrão está configurado como 1.0005 amu (unidades de massa atômica). Um valor típico é 1.0 amu para dados de espectrômetros de baixa resolução. Valores menores, como 0.02 amu, são usados para dados de alta resolução. O Fragment bin offset (Deslocamento da massa dos fragmentos) possui o valor 0.4 referente à uma pequena correção que permite ajustar o centro da janela de tolerância, útil para compensar pequenos desvios na calibração do instrumento (CARVALHO et al., 2008; CARVALHO et al., 2009; ENG; JAHAN; HOOPMANN, 2013;SANTOS et al., 2022; CLASEN et al., 2023).
-
Clear mz range start/stop (Limpar intervalo de massas m/z): exclui determinados intervalos de massas do espectro MS/MS da análise, por exemplo, para remover sinais de reagentes de marcação como iTRAQ ou TMT. Para o valor comum, nenhum intervalo está configurado, o que significa que todos os sinais serão usados na busca (CARVALHO et al., 2008; CARVALHO et al., 2009; ENG; JAHAN; HOOPMANN, 2013;SANTOS et al., 2022; CLASEN et al., 2023).
-
Search mass range start/stop (Intervalo de massas a ser pesquisado): especifica o intervalo de massas dos íons precursores a serem pesquisados. Os valores comuns estão configurados entre 500 a 6000 amu, indicando que apenas peptídeos dentro desse intervalo de massas serão considerados. Uma dica é ajustar esse valor de acordo com o tamanho dos peptídeos que você espera. Para proteínas maiores ou marcadores de proteínas, pode ser necessário ajustar o intervalo (CARVALHO et al., 2008; CARVALHO et al., 2009; ENG; JAHAN; HOOPMANN, 2013;SANTOS et al., 2022; CLASEN et al., 2023).
-
Max variable mods per peptide (Máximo de modificações variáveis por peptídeo): define o número máximo de modificações pós-traducionais (PTMs) que podem ser aplicadas a cada peptídeo durante a busca. Isso é útil para incluir modificações como fosforilação, oxidação de metionina, entre outras. Por exemplo, se definido para 2, o Comet irá considerar até 2 modificações por peptídeo (CARVALHO et al., 2008; CARVALHO et al., 2009; ENG; JAHAN; HOOPMANN, 2013;SANTOS et al., 2022; CLASEN et al., 2023).
-
Missed cleavages (Clivagens enzimáticas perdidas): permite especificar quantas clivagens enzimáticas perdidas são aceitas. Quando a enzima não consegue cortar em todos os locais esperados (por exemplo, tripsina não corta todos os resíduos de lisina ou arginina), essas clivagens não realizadas são chamadas de “missed cleavages”. O padrão para a tripsina é permitir até 2 clivagens perdidas, mas isso pode ser ajustado de acordo com o seu experimento (CARVALHO et al., 2008; CARVALHO et al., 2009; ENG; JAHAN; HOOPMANN, 2013;SANTOS et al., 2022; CLASEN et al., 2023).
-
Theoretical Fragment Ions (Íons fragmentos teóricos): especifica quais tipos de íons de fragmentos teóricos serão usados para a comparação com os espectros MS/MS. Os Valores comuns pode ser: M peak only que, por padrão, está configurado para usar apenas íons do tipo B e Y, que são os mais comuns em espectros de MS/MS; Neutral Loss que está habilitado, o que significa que o software também levará em conta a perda neutra de fragmentos, que ocorre quando moléculas como água ou amônia são eliminadas do peptídeo (CARVALHO et al., 2008; CARVALHO et al., 2009; ENG; JAHAN; HOOPMANN, 2013;SANTOS et al., 2022; CLASEN et al., 2023).
-
Enzyme (Enzima): define a enzima usada para digerir as proteínas no experimento. No exemplo, está configurado para Trypsin/P, uma forma modificada da tripsina que cliva após resíduos de lisina (K) ou arginina (R). Se estiver usando outra enzima, como Lys-C, certifique-se de selecioná-la aqui. Enzimas específicas podem ser selecionadas dependendo do protocolo experimental (CARVALHO et al., 2008; CARVALHO et al., 2009; ENG; JAHAN; HOOPMANN, 2013;SANTOS et al., 2022; CLASEN et al., 2023).
-
Enzyme specificity (Especificidade enzimática): define a especificidade enzimática. Aqui está ajustado para Semi-specific, o que significa que a enzima pode cortar em um ou ambos os lados do peptídeo. Para digestão completa, use a opção Full-specific, que exige clivagem em ambos os lados (CARVALHO et al., 2008; CARVALHO et al., 2009; ENG; JAHAN; HOOPMANN, 2013;SANTOS et al., 2022; CLASEN et al., 2023).
-
Homo sapiens PEFF search (Busca no banco PEFF): habilita a busca em bancos de dados PEFF (Protein Extended FASTA Format), que são usados para identificar variantes de proteínas humanas. Por padrão, a opção está desabilitada, o que significa que o banco de dados PEFF não está sendo utilizado (CARVALHO et al., 2008; CARVALHO et al., 2009; ENG; JAHAN; HOOPMANN, 2013;SANTOS et al., 2022; CLASEN et al., 2023).
-
N15 Labeling (Rotulagem com N15): indica se o experimento usa rotulagem isotópica com Nitrogênio-15 (N15), que é uma técnica de quantificação baseada em isótopos. Por padrão, não está habilitado, indicando que a rotulagem isotópica não foi aplicada no experimento (CARVALHO et al., 2008; CARVALHO et al., 2009; ENG; JAHAN; HOOPMANN, 2013; SANTOS et al., 2022; CLASEN et al., 2023).
Os parâmetros exibidos na aba “Search and Filtering” permite ajustar de maneira precisa os parâmetros para a identificação de peptídeos e proteínas a partir de dados de espectrometria de massa (Ver Figura 7).
Com a combinação de filtros de qualidade e estimativas de taxa de falso positivo, é possível maximizar a acurácia e confiabilidade dos resultados.
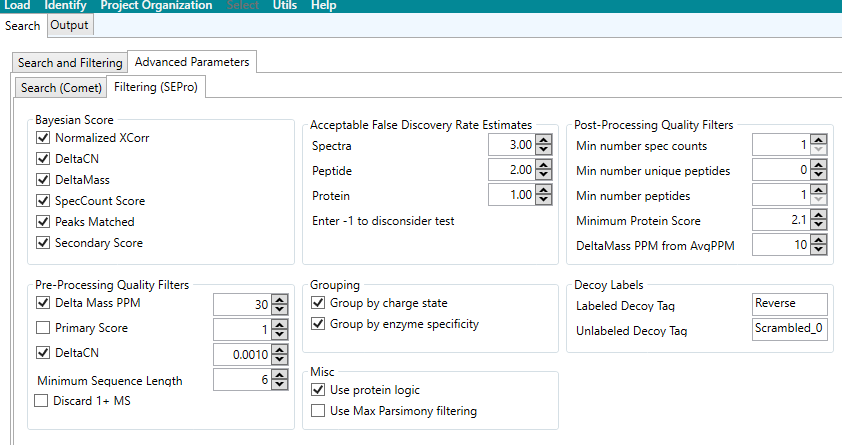 Figura 7 – Parâmetros avançados presentes na opção “Search and Filtering” do menu “Identify”.
Figura 7 – Parâmetros avançados presentes na opção “Search and Filtering” do menu “Identify”.
A seguir, será fornecida uma explicação detalhada de cada parâmetro, destacando suas funcionalidades e apresentando exemplos práticos de aplicação.
-
Bayesian Score (Pontuação Bayesiana)
-
Normalized XCorr: Utiliza o XCorr (cross-correlation score) normalizado para pontuar a correspondência do espectro. Este valor compara a similaridade do espectro experimental com o espectro teórico do peptídeo (CARVALHO et al., 2012).
-
DeltaMass: Habilitado para filtrar espectros com base na diferença entre a massa medida e a massa teórica calculada do peptídeo (CARVALHO et al., 2012).
-
SpecCount Score: Usa a contagem de espectros como um parâmetro adicional para quantificar a confiança na identificação de um peptídeo (CARVALHO et al., 2012).
-
Peaks Matched: Considera o número de picos que coincidem entre os espectros medidos e os esperados (CARVALHO et al., 2012).
-
Secondary Score: Usa uma pontuação secundária para melhorar a confiança na identificação (CARVALHO et al., 2012).
-
-
Acceptable False Discovery Rate Estimates (Estimativas aceitáveis da taxa de descoberta falsa)
-
Spectra: Está configurado para 3.00, o que significa que a taxa de falsos positivos para espectros de peptídeos pode ser de até 3% (CARVALHO et al., 2012).
-
Peptide: Configurado como 2.00, limitando a taxa de falsos positivos na identificação de peptídeos (CARVALHO et al., 2012).
-
Protein: Configurado para 1.00, garantindo um rigor mais alto na identificação de proteínas (CARVALHO et al., 2012).
-
Enter 1-to-Disconsider Test: Define a desconsideração de pontuações abaixo de 1 (CARVALHO et al., 2012).
-
-
Post-Processing Quality Filters (Filtros de qualidade pós-processamento)
-
Min number spec counts: Define o número mínimo de espectros que um peptídeo deve ter para ser considerado válido. Aqui está ajustado para 1 (CARVALHO et al., 2012).
-
Min number unique peptides: Define o número mínimo de peptídeos únicos para identificar uma proteína. Está em 0, o que significa que não há restrição de peptídeos únicos (CARVALHO et al., 2012).
-
Min number peptides: Exige pelo menos 1 peptídeo para confirmar a presença de uma proteína (CARVALHO et al., 2012).
-
Minimum Protein Score: Configurado como 2.1, o que significa que a pontuação mínima necessária para identificar uma proteína é 2.1 (CARVALHO et al., 2012).
-
DeltaMass PPM from AvgPPM: Configurado para 10 ppm, definindo a variação máxima permitida entre as massas observadas e as esperadas após o processamento (CARVALHO et al., 2012).
-
-
Pre-Processing Quality Filters (Filtros de qualidade no pré-processamento)
-
Delta Mass PPM: Define a precisão de massa permitida entre a massa observada e a teórica antes do processamento, configurada para 30 ppm (CARVALHO et al., 2012).
-
Primary Score: Usa uma pontuação primária para filtrar peptídeos com base em suas correspondências (CARVALHO et al., 2012).
-
DeltaCN: Configurado como 0.0010, esta métrica avalia a diferença na pontuação entre o melhor e o segundo melhor candidato a peptídeo (CARVALHO et al., 2012).
-
Minimum Sequence Length: Define o comprimento mínimo da sequência de peptídeos que será considerada, configurado como 6 aminoácidos (CARVALHO et al., 2012).
-
Discard 1+ MS: quando não está habilitado, significa que peptídeos com uma única carga também serão considerados (CARVALHO et al., 2012).
-
-
Grouping (Agrupamento)
-
Group by charge state: Agrupa os peptídeos com base no estado de carga (CARVALHO et al., 2012).
-
Group by enzyme specificity: Agrupa peptídeos de acordo com a especificidade enzimática utilizada na digestão proteica (CARVALHO et al., 2012).
-
-
Decoy Labels (Rótulos para banco “decoy”)
-
Labeled Decoy Tag: Configurado como Reverse, indicando que o banco “decoy” (controle negativo) é gerado revertendo a sequência proteica (CARVALHO et al., 2012).
-
Unlabeled Decoy Tag: Configurado como Scrambled_0, indicando que as sequências são embaralhadas para gerar o banco “decoy” (CARVALHO et al., 2012).
-
-
Miscellaneous (Miscelânea)
-
Use protein logic: Utiliza a lógica de proteínas ao identificar conjuntos de peptídeos (CARVALHO et al., 2012).
-
Use Max Parsimony filtering: Habilita a filtragem de máxima parcimônia, priorizando a identificação mínima de proteínas para explicar os peptídeos observados (CARVALHO et al., 2012).
-
Um aspecto interessante é que a ferramenta possui, no menu “Help“, um fórum que permite o contato direto entre pesquisadores e o coordenador do laboratório. Isso garante que a ferramenta esteja constantemente atualizada, com base nas contribuições e feedbacks dos usuários.
PREPARAÇÃO DE DADOS
Na análise proteômica, o uso de bases de dados confiáveis é fundamental para garantir a precisão na identificação de proteínas e suas variantes. Aqui, utilizaremos duas fontes principais para a preparação dos dados: bases de dados de proteínas de referência e dados de espectrometria de massa provenientes de pacientes. Além disso, criaremos uma terceira base de dados de proteínas contendo mutações para aumentar o número de proteínas identificadas.
Para identificar e anotar proteínas humanas em amostras biológicas, utilizaremos o banco de dados UniProt. A plataforma UniProt fornece sequências de proteínas revisadas e anotadas, que são essenciais para garantir uma correspondência precisa durante a análise proteômica. Esse banco de dados é amplamente utilizado na comunidade científica por seu rigor na curadoria de dados, o que facilita o processo de busca de peptídeos em ferramentas como o PatternLab for Proteomics. A base de dados escolhida pode ser baixada diretamente da UniProt, e inclui informações tanto de proteínas revisadas quanto de variantes reportadas, permitindo uma análise mais abrangente.
Além do banco de dados de proteínas, será utilizado um conjunto de dados de espectrometria de massa de pacientes com câncer, disponível no Proteomics Data Commons (PDC 000111). Esses dados fornecem perfis proteômicos detalhados de amostras tumorais, que são cruciais para a correlação com estados patológicos e o entendimento da biologia subjacente ao câncer. A combinação desses dados de espectrometria de massa com as sequências de referência da UniProt permite uma análise mais aprofundada, focada na identificação de variantes de peptídeos relacionadas a mutações, frequentemente encontradas em pacientes oncológicos. A seguir, estão detalhadas as etapas realizadas para a preparação dos dados.
-
Download de Sequências de Proteínas: O primeiro passo é baixar as sequências proteicas relevantes do banco de dados UniProt, assegurando-se de incluir tanto as proteínas revisadas quanto aquelas contendo variantes. Isso possibilita uma busca mais ampla e detalhada por peptídeos mutados.
-
Integração com Dados de Espectrometria: Em seguida, os dados de espectrometria de massa provenientes do Proteomics Data Commons (THANGUDU et al., 2022) serão carregados no PatternLab for Proteomics. Para exemplificar, utilizamos a amostra TCGA-A6-3810-01A. A combinação desses dados espectrométricos com as sequências de referência facilitará a identificação de proteínas expressas, incluindo potenciais variantes associadas ao câncer.
-
Filtragem e Formatação: Com as bases de dados carregadas, é necessário aplicar filtros específicos no PatternLab para eliminar entradas redundantes ou não relevantes e formatar os dados para a análise downstream. A formatação adequada é essencial para garantir a qualidade dos resultados, especialmente em estudos de larga escala que envolvem dados clínicos e experimentais complexos.
Construção da Base de Dados de proteínas variantes
Para construir o banco de dados de proteínas variantes, foram seguidos os procedimentos metodológicos descritos no trabalho de Cunha et al. (2022). Para isso, foram coletados dados da RefSeq, que fornece sequências referenciais de proteínas, e do dbSNP, que contém informações de variações de nucleotídeos únicos (SNPs). Ambas as bases de dados estão disponíveis no portal NCBI, acessíveis pelos links: (https://ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/mRNA_Prot/) e (https://ftp.ncbi.nlm.nih.gov/snp/). Esses dados incluem informações sobre variantes genéticas, como mutações de nucleotídeos e alterações nas sequências de aminoácidos. O formato dos dados brutos pode conter uma grande variedade de mutações. Contudo, para este trabalho, serão abordadas exclusivamente as mutações de substituição, onde ocorre a troca de aminoácidos. Dessa forma, surge a necessidade de pré-processar o banco de dados de variantes, facilitando a visualização e o manejo do código, considerando o padrão HGVS (Human Genome Variation Society).
Nomenclatura HGVS
A nomenclatura HGVS (Human Genome Variation Society) é amplamente reconhecida para a descrição de mutações em proteínas e DNA, proporcionando um padrão unificado que facilita a interpretação das variações genéticas. O formato utilizado neste trabalho para descrever trocas de aminoácidos segue esse padrão, conforme ilustra a Figura 8.
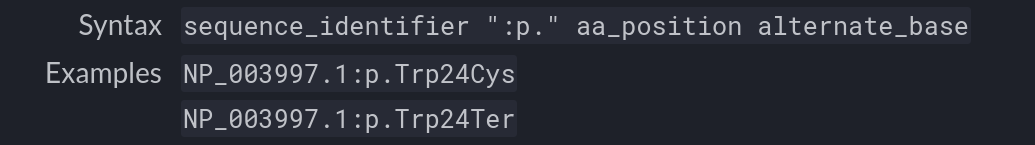 Figura 8 – Exemplo de nomenclatura HGVS. 2024. Disponível em: Substitution – HGVS Nomenclature.
Figura 8 – Exemplo de nomenclatura HGVS. 2024. Disponível em: Substitution – HGVS Nomenclature.
Nessa sintaxe o sequence_identifier representa o ID da proteína, p. indica que a mutação ocorre a nível de proteína, aa_position refere-se à posição do aminoácido na sequência, e alternate_base identifica o aminoácido substituto.
Por exemplo: NP_003997.1:p.Trp24Cys indica uma mutação na proteína identificada por NP_003997.1, onde o aminoácido triptofano (Trp) na posição 24 foi substituído por cisteína (Cys).
Pré-Processamento do arquivo
O processamento do arquivo envolve a divisão de cada linha em colunas, extraindo o ID da proteína, a mutação (SNP), o aminoácido de referência, a posição da mutação e o aminoácido mutado, conforme demonstrado na Figura 9.
Para gerar o banco de dados, é necessário desenvolver um script na linguagem de preferência, que processe dois arquivos de entrada: um contendo as sequências de proteínas (RefSeq) e outro com informações sobre mutações (SNP, previamente processado). O objetivo é modificar as sequências de proteínas com base nas mutações identificadas, gerar peptídeos mutados e salvar os resultados em um arquivo de saída.
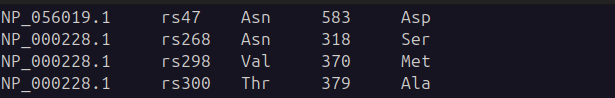 Figura 9 – Exemplo de arquivo filtrado esperado.
Figura 9 – Exemplo de arquivo filtrado esperado.
Descrição da lógica de programação
A lógica de programação do script inicia verificando os parâmetros de entrada para garantir que os arquivos corretos sejam fornecidos. Em seguida, as sequências de proteínas são lidas e armazenadas em uma estrutura de dados do tipo hash, associando cada ID de proteína (chave) à sua respectiva sequência de aminoácidos (valor). O arquivo de mutações é então processado, aplicando substituições de aminoácidos nas posições específicas das proteínas.
As sequências modificadas são divididas em peptídeos trípticos, fragmentos que se formam nas clivagens de lisina (K) e arginina (R). Apenas peptídeos com tamanho entre 7 e 35 aminoácidos são considerados válidos. Se uma mutação resulta em um códon de parada, a sequência é truncada até essa posição. Finalmente, os peptídeos mutados válidos são concatenados e gravados no arquivo final, representando as sequências proteicas modificadas com suas respectivas mutações.
Leitura e Armazenamento de Sequências
A primeira etapa envolve a leitura do arquivo de proteínas. Cada sequência de proteína é identificada por um ID único (RefSeq ID), e as respectivas sequências de aminoácidos são armazenadas para processamento posterior.
INICIAR leitura do arquivo de proteínas
PARA cada linha no arquivo de proteínas
SE a linha inicia com ">"
IDENTIFICAR o ID da proteína
SENÃO
ADICIONAR à sequência ao hash de proteínas usando o ID
FIM da leitura do arquivo de proteínas
Aplicação de Mutações às Sequências de Proteínas
Para cada linha do arquivo de mutações, os seguintes elementos são extraídos:
-
ID da proteína: Identifica a proteína cuja sequência será modificada.
-
Aminoácido de referência (antes da mutação): O aminoácido presente na sequência original da proteína.
-
Posição da mutação: A posição onde a mutação ocorre.
-
Aminoácido mutado: O aminoácido que substituirá o de referência.
Para cada mutação descrita, verifica-se se o ID da proteína correspondente está presente no hash de proteínas. Se encontrado, a mutação é aplicada à sequência de aminoácidos.
Pseudocódigo:
INICIAR leitura do arquivo de mutações
PARA cada linha no arquivo de mutações
EXTRAIR o ID da proteína, posição, aminoácido de referência e aminoácido mutado
SE o ID da proteína está no hash de proteínas
SUBSTITUIR o aminoácido de referência pelo aminoácido mutado na sequência
SENÃO
CONTINUAR para a próxima linha
FIM da leitura do arquivo de mutações
Verificação de Condições para o Peptídeo
Após aplicar as mutações às sequências de aminoácidos, é necessário verificar se os fragmentos gerados são válidos de acordo com os seguintes critérios:
-
Criação de peptídeos trípticos: As sequências são fragmentadas em peptídeos trípticos, ou seja, pedaços gerados pela clivagem nos resíduos de R (arginina) e K (lisina). Cada fragmento corresponde a uma substring entre duas dessas clivagens ou até o fim da sequência.
-
Tamanho dos peptídeos: Apenas peptídeos que possuem entre 7 e 35 aminoácidos são considerados válidos. Peptídeos menores ou maiores são descartados, pois não atendem aos critérios estabelecidos para tamanho ideal de peptídeos mutados.
-
Mutações de parada: Se a mutação resultar em um códon de parada (representado pelo aminoácido Ter), a sequência é truncada até o ponto onde ocorre a parada. O objetivo é gerar fragmentos de peptídeos que parem na posição correta da mutação.
Geração do Arquivo Final
O arquivo final, contém as sequências mutadas válidas para cada proteína. O formato do arquivo segue o padrão de FASTA, onde o ID da proteína é seguido pelas suas sequências mutadas concatenadas.
Pseudocódigo:
INICIAR escrita do arquivo final
PARA cada ID de proteína processado
SE o ID mudou em relação à linha anterior
ESCREVER as sequências concatenadas da proteína anterior
CONCATENAR novos fragmentos para o ID atual
FIM da escrita do arquivo final
ANÁLISE DE CORRESPONDÊNCIA DE ESPECTRO DE PEPTÍDEOS
A análise de correspondência de espectro de peptídeos é uma etapa crítica na identificação de proteínas em experimentos de proteômica. Esse processo envolve a comparação dos espectros de massa obtidos experimentalmente com espectros teóricos gerados a partir de sequências de proteínas de um banco de dados. Para garantir a precisão da correspondência, é fundamental inserir corretamente os parâmetros experimentais utilizados na coleta dos dados, pois a ferramenta de busca simula as condições do experimento com base nesses parâmetros.
Para o estudo PDC 000111, os seguintes parâmetros experimentais foram utilizados:
-
Estratégia de Quantificação: A quantificação foi realizada utilizando a abordagem Label-free, que não requer a marcação química dos peptídeos, mas sim a comparação direta das intensidades dos sinais nos espectros de massa.
-
Enzima Digestiva: A enzima escolhida para a digestão proteica foi a tripsina, que cliva as proteínas preferencialmente nas ligações peptídicas após resíduos de lisina e arginina, gerando peptídeos de tamanhos ideais para a análise por espectrometria de massa.
-
Reagente de Alquilação: Foi utilizado iodoacetamida como reagente de alquilação, que reage com resíduos de cisteína para evitar a formação de pontes dissulfeto e estabilizar a estrutura das proteínas.
-
Instrumento de Espectrometria de Massa: O instrumento usado para coletar os espectros de massa foi o Orbitrap Velos, um espectrômetro de alta resolução amplamente utilizado para análises proteômicas devido à sua precisão na determinação de massas e eficiência na análise de amostras complexas.
A Figura 10 mostra a ferramenta com os parâmetros selecionados, junto aos dados experimentais e à base de dados teórica. Os demais parâmetros permaneceram nos valores padrão, conforme as recomendações de Carvalho et al. (2009) e Santos et al. (2022).
 Figura 10 – Parâmetros utilizados para análise proteômica de câncer de colorretal.
Figura 10 – Parâmetros utilizados para análise proteômica de câncer de colorretal.
FILTROS ESTATÍSTICOS E ORGANIZAÇÃO DOS DADOS
Os resultados da análise utilizando o PatternLab for Proteomics mostraram uma alta confiabilidade nas identificações de espectros, peptídeos e proteínas (Ver Figura 11). Na parte superior da tabela, é possível observar que a taxa de falso positivo para espectros (Spec FDR) foi de 0,62%, com 21 espectros classificados como falsos positivos entre os 3.378 espectros analisados. Para os peptídeos, a taxa de falso positivo (Pep FDR) foi ainda menor, de 0,21%, com apenas 4 dos 1.875 peptídeos identificados sendo considerados falsos positivos. No nível das proteínas, a taxa de falso positivo (Prot FDR) foi de 0,37%, com 4 proteínas identificadas incorretamente entre as 1.092 analisadas.
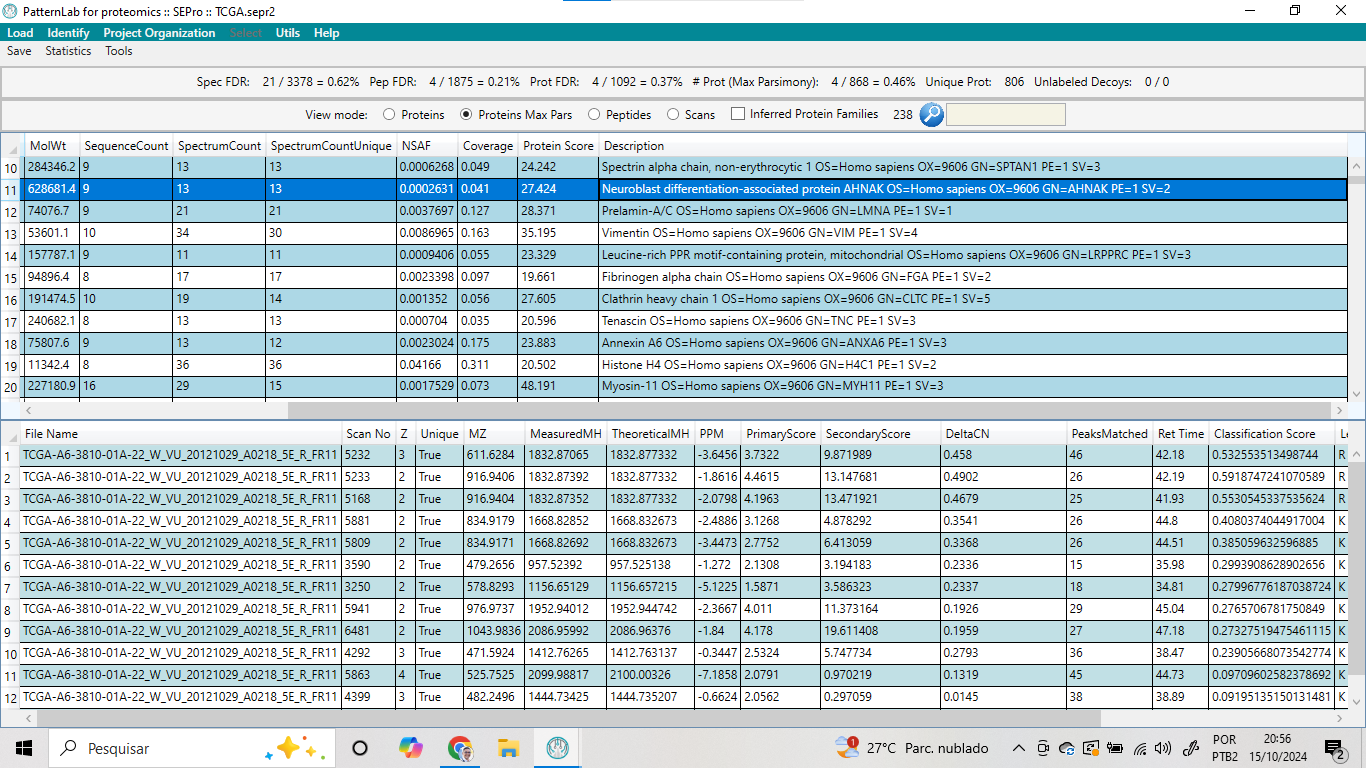 Figura 11 – Resultado da busca utilizando o banco de dados de proteínas presente no UNIPROT.
Figura 11 – Resultado da busca utilizando o banco de dados de proteínas presente no UNIPROT.
Quando a análise foi realizada no modo de máxima parcimônia, 868 proteínas foram identificadas, e a taxa de falso positivo para proteínas neste modo foi de 0,46%. Além disso, foram identificadas 806 proteínas únicas, o que evidencia a diversidade de proteínas presentes na amostra e a minimização de redundâncias. Não foram observados “decoys” não rotulados, confirmando a ausência de identificações incorretas provenientes de sequências aleatórias utilizadas como controle negativo.
Nos resultados obtidos pela análise proteômica, a proteína Neuroblast differentiation-associated protein (AHNAK), com peso molecular de 628.681,4 Da, foi identificada com base em 13 espectros únicos, correspondentes a 9 sequências de peptídeos. A abundância relativa da proteína, expressa pelo NSAF (Normalized Spectral Abundance Factor), foi de 0,0006321, indicando uma baixa quantidade relativa na amostra. A cobertura da sequência da proteína foi de 12,7%, o que sugere que uma parte significativa da proteína foi detectada na análise. A pontuação da proteína (Protein Score), de 27.424, reforça a confiabilidade da identificação.
O peptídeo destacado, proveniente do espectro de número 5232, apresentou uma carga de 3 e foi considerado único para a proteína AHNAK. A razão massa/carga (m/z) medida foi de 611,6284, e a massa do íon peptídico, tanto medida quanto teórica, coincidiu exatamente, com um erro de massa de -3,6456 ppm, confirmando a precisão da identificação. A correspondência entre o espectro e a sequência peptídica foi corroborada por uma pontuação primária de 3,7322 e uma pontuação secundária de 8,971899, com 46 picos do espectro casados com fragmentos do peptídeo. Esses resultados indicam uma identificação robusta da proteína AHNAK na amostra analisada.
O mesmo procedimento de correspondência de espectro foi realizado utilizando o banco de dados de proteínas mutadas. No entanto, por se tratarem de bancos de dados distintos, a busca deve ser conduzida separadamente para proteínas de referência e proteínas mutadas. Isso ocorre porque a inclusão de proteínas mutadas no mesmo banco de dados de referência aumenta significativamente o espaço de busca.
Em algumas abordagens, tanto as proteínas mutadas quanto as de referência são combinadas em um único arquivo de busca. Embora essa prática possa parecer eficiente, ela apresenta um grande risco: o aumento do espaço de busca leva a um número maior de correspondências potenciais, o que pode resultar em um aumento nos falsos positivos (CUNHA et al., 2022). Isso torna mais difícil identificar com precisão peptídeos variantes e introduz incertezas nos resultados. Para mitigar esse problema, é recomendável separar as buscas ou adotar técnicas de filtragem rigorosas que minimizem a inclusão de resultados errôneos e garantam maior confiança nas identificações realizadas.
Os dados apresentados na tabela foram obtidos utilizando um banco de dados de proteínas mutadas, com o objetivo de identificar peptídeos mutados em uma amostra. A análise demonstra uma alta qualidade nas identificações realizadas, conforme evidenciado pelos índices de descoberta falsa (FDR) apresentados na parte superior da tabela. O Spec FDR, que representa a taxa de descoberta falsa para espectros, foi de 0.15% (5/3351), enquanto o Pep FDR, relativo aos peptídeos, foi de 0.28% (7/2491). Esses valores indicam que há uma confiança considerável nas identificações de espectros e peptídeos, com baixas taxas de falsos positivos. Além disso, a taxa de descoberta falsa para proteínas (Prot FDR) foi de 0.46% (4/869), reforçando a confiabilidade na identificação das proteínas presentes na amostra. No total, foram identificadas 356 proteínas únicas, um número significativo que denota a abrangência da análise.
Na linha selecionada da tabela, referente à proteína “proliferation-associated protein 2G4 [Homo sapiens]”, observamos que o número de peptídeos únicos identificados foi 5, com um valor de NSAF (Normalized Spectral Abundance Factor) de 0.0015487, que fornece uma estimativa relativa da abundância da proteína. O Protein Score de 5.076 reflete a qualidade da identificação da proteína, e a cobertura de 5.6% indica a porção da sequência total de aminoácidos identificada no experimento.
No que diz respeito à qualidade dos espectros, a tabela inferior mostra parâmetros como a diferença entre o valor observado e o teórico de massa/razão de carga (PPM), com valores negativos indicando uma correspondência próxima entre o espectro observado e o calculado. Para a proteína em questão, o valor de PPM variou entre -5.7361 e -1.2081, sugerindo uma precisão alta. A classificação dos peptídeos, expressa pelo Classification Score, variou de 0.2527 a 0.3668, o que reforça a confiança na correta atribuição dos peptídeos à proteína mutada.
Ao utilizar bancos de dados distintos para buscar peptídeos a partir de um mesmo experimento, é fundamental comparar a qualidade da identificação dos peptídeos identificados sob o mesmo número de scan. Nesse contexto, a análise dos scores de cada correspondência é crucial. O trabalho de Cunha et al. estabelece que, ao selecionar peptídeos redundantes, é necessário considerar aqueles com scores que apresentem uma margem superior a 20%. Essa abordagem assegura que a escolha seja fundamentada em evidências robustas, minimizando a probabilidade de falsos positivos. Essa prática é especialmente relevante em estudos proteômicos, onde a identificação precisa de peptídeos é vital para a interpretação biológica dos dados, especialmente em contextos clínicos, como na análise de amostras de pacientes com câncer, onde a precisão pode impactar diretamente o diagnóstico e o tratamento.
O Quadro 1 apresenta os peptídeos mutados, seus respectivos identificadores de proteínas e SNP IDs, além das substituições de aminoácidos correspondentes. É importante ressaltar que os dados apresentados servem apenas como exemplo de como a identificação de peptídeos mutados pode ser realizada. No entanto, para garantir a precisão dos resultados, é fundamental realizar a validação por meio da comparação dos scores dos peptídeos identificados sob o mesmo número de scan com os peptídeos contidos no banco de dados de referência. Essa validação é crucial para assegurar a confiabilidade das identificações e para minimizar a possibilidade de erros, como falsos positivos, na análise proteômica.
Quadro 1 – Peptídeos mutados identificados em uma amostra de câncer de colorretal.
|
RefSeq ID |
SNP ID |
Protein Change |
Peptideo Referência |
Peptideo Mutado |
|
NP_056019 |
rs47 |
p.Asn583Asp |
LGnCEPDNGK |
LGdCEPDNGK |
|
NP_006073 |
rs1059274 |
p.Ser340Thr |
sIQFVDWCPTGFK |
tIQFVDWCPTGFK |
|
NP_997195 |
rs2261398 |
p.Ser162Gly |
LSVDYsK |
LSVDYgK |
|
NP_000509 |
rs34831026 |
p.Arg41Lys |
LLVVYPWTQrFFESFGDLSTPDAVMGNPK |
FFESFGDLSTPDAVMGNPK |
|
NP_000510 |
rs35395083 |
p.Ala23Glu |
VNVDaVGGEALGR |
VNVDeVGGEALGR |
|
NP_002107 |
rs45468091 |
p.Lys145Arg |
QDAYDGkDYIALNEDLR |
DYIALNEDLR |
|
NP_001229687 |
rs45468091 |
p.Lys145Arg |
QDAYDGkDYIALNEDLR |
DYIALNEDLR |
|
NP_001806 |
rs61737016 |
p.Ala93Pro |
VDGNSLIVGYVIGTQQATPGaAYSGR |
VDGNSLIVGYVIGTQQATPGpAYSGR |
|
NP_001264092 |
rs61737016 |
p.Ala93Pro |
VDGNSLIVGYVIGTQQATPGaAYSGR |
VDGNSLIVGYVIGTQQATPGpAYSGR |
|
NP_002147 |
rs61755731 |
p.Arg142Lys |
rGVMLAVDAVIAELK |
GVMLAVDAVIAELK |
|
NP_955472 |
rs61755731 |
p.Arg142Lys |
rGVMLAVDAVIAELK |
GVMLAVDAVIAELK |
|
NP_001264012 |
rs71221348 |
p.Cys700Arg |
AVFPSIVGcPR |
AVFPSIVGr |
|
NP_112420 |
rs78587033 |
p.Lys106Arg |
kIFVGGIK |
IFVGGIK |
|
NP_002127 |
rs78587033 |
p.Lys106Arg |
kIFVGGIK |
IFVGGIK |
|
NP_001327 |
rs140637018 |
p.Arg189Lys |
NYTLWrVGDYGSLSGR |
VGDYGSLSGR |
|
NP_001113 |
rs140787521 |
p.Arg93Lys |
IEErLPILNQPSTQIVANAK |
LPILNQPSTQIVANAK |
|
NP_001226 |
rs142663000 |
p.Lys308Arg |
AVAISLPkGVVEVTHDLQK |
GVVEVTHDLQK |
|
NP_001193943 |
rs142663000 |
p.Lys308Arg |
AVAISLPkGVVEVTHDLQK |
GVVEVTHDLQK |
|
NP_004478 |
rs143141179 |
p.Lys1424Arg |
EAALTkIQTEIIEQEDLIK |
IQTEIIEQEDLIK |
|
NP_001243417 |
rs143141179 |
p.Lys1349Arg |
EAALTkIQTEIIEQEDLIK |
IQTEIIEQEDLIK |
|
NP_001243416 |
rs143141179 |
p.Lys1390Arg |
EAALTkIQTEIIEQEDLIK |
IQTEIIEQEDLIK |
|
NP_001243415 |
rs143141179 |
p.Lys1429Arg |
EAALTkIQTEIIEQEDLIK |
IQTEIIEQEDLIK |
|
NP_000081 |
rs144614075 |
p.Lys1273Arg |
FCHPELkSGEYWVDPNQGCK |
SGEYWVDPNQGCK |
|
NP_612398 |
rs151128422 |
p.Lys221Arg |
GMLSAITNVVQNTGkSVLTGGLDALEFIGK |
SVLTGGLDALEFIGK |
|
NP_001317693 |
rs151128422 |
p.Lys14Arg |
MLSAITNVVQNTGkSVLTGGLDALEFIGK |
SVLTGGLDALEFIGK |
|
NP_001317693 |
rs151128422 |
p.Lys14Arg |
MLSAITNVVQNTGkSVLTGGLDALEFIGK |
SVLTGGLDALEFIGK |
|
NP_001265281 |
rs189552242 |
p.Lys644Arg |
STYIkSPPFFENLTLDLQPPK |
SPPFFENLTLDLQPPK |
|
NP_002188 |
rs189552242 |
p.Lys644Arg |
STYIkSPPFFENLTLDLQPPK |
SPPFFENLTLDLQPPK |
|
NP_005568 |
rs199684452 |
p.Glu1743Asp |
ATTVTGTPCQeWAAQEPHR |
ATTVTGTPCQdWAAQEPHR |
|
NP_001158886 |
rs200332813 |
p.Lys119Arg |
DYNVTANSkLVIITAGAR |
LVIITAGAR |
|
NP_001158888 |
rs200332813 |
p.Lys90Arg |
DYNVTANSkLVIITAGAR |
LVIITAGAR |
|
NP_001158887 |
rs200332813 |
p.Lys90Arg |
DYNVTANSkLVIITAGAR |
LVIITAGAR |
|
NP_005557 |
rs200332813 |
p.Lys90Arg |
DYNVTANSkLVIITAGAR |
LVIITAGAR |
|
NP_001307148 |
rs200396401 |
p.Lys621Arg |
FFETkLCQIFSDLNATYR |
LCQIFSDLNATYR |
|
NP_001307151 |
rs200396401 |
p.Lys189Arg |
FFETkLCQIFSDLNATYR |
LCQIFSDLNATYR |
|
NP_001307149 |
rs200396401 |
p.Lys213Arg |
FFETkLCQIFSDLNATYR |
LCQIFSDLNATYR |
|
NP_001073884 |
rs200396401 |
p.Lys622Arg |
FFETkLCQIFSDLNATYR |
LCQIFSDLNATYR |
|
NP_001231825 |
rs201083895 |
p.Lys336Arg |
ATLSkHQAVQLPR |
HQAVQLPR |
|
NP_001231826 |
rs201083895 |
p.Lys403Arg |
ATLSkHQAVQLPR |
HQAVQLPR |
|
NP_001231827 |
rs201083895 |
p.Lys437Arg |
ATLSkHQAVQLPR |
HQAVQLPR |
|
NP_001157260 |
rs201083895 |
p.Lys403Arg |
ATLSkHQAVQLPR |
HQAVQLPR |
|
NP_001157262 |
rs201083895 |
p.Lys434Arg |
ATLSkHQAVQLPR |
HQAVQLPR |
|
NP_005147 |
rs201083895 |
p.Lys431Arg |
ATLSkHQAVQLPR |
HQAVQLPR |
|
NP_001229971 |
rs281860474 |
p.Lys145Arg |
GYDQSAYDGkDYIALNEDLR |
DYIALNEDLR |
|
NP_002108 |
rs281860474 |
p.Lys145Arg |
GYDQSAYDGkDYIALNEDLR |
DYIALNEDLR |
|
NP_005505 |
rs281864619 |
p.Lys145Arg |
GHDQYAYDGkDYIALNEDLR |
DYIALNEDLR |
|
NP_005960 |
rs369030699 |
p.Lys83Arg |
CAHIEAkFYEEVHDLER |
FYEEVHDLER |
|
NP_005381 |
rs369861495 |
p.Gly169Val |
NFYGGNGIVGAQgPLGAGIALACK |
NFYGGNGIVGAQvPLGAGIALACK |
|
NP_000509 |
rs369865419 |
p.Lys18Arg |
SAVTALWGkVNVDEVGGEALGR |
VNVDEVGGEALGR |
VISUALIZAÇÃO DE DADOS
No que diz respeito à visualização de dados, o PatternLab for Proteomics oferece ao usuário a funcionalidade de visualizar o gráfico do espectro de massas para cada peptídeo identificado. Para acessar essa visualização, basta clicar duas vezes sobre o espectro desejado, e o gráfico correspondente será exibido (ver Figura 12). Essa facilidade de visualização é essencial para a interpretação dos dados proteômicos, permitindo uma análise mais detalhada dos resultados obtidos.
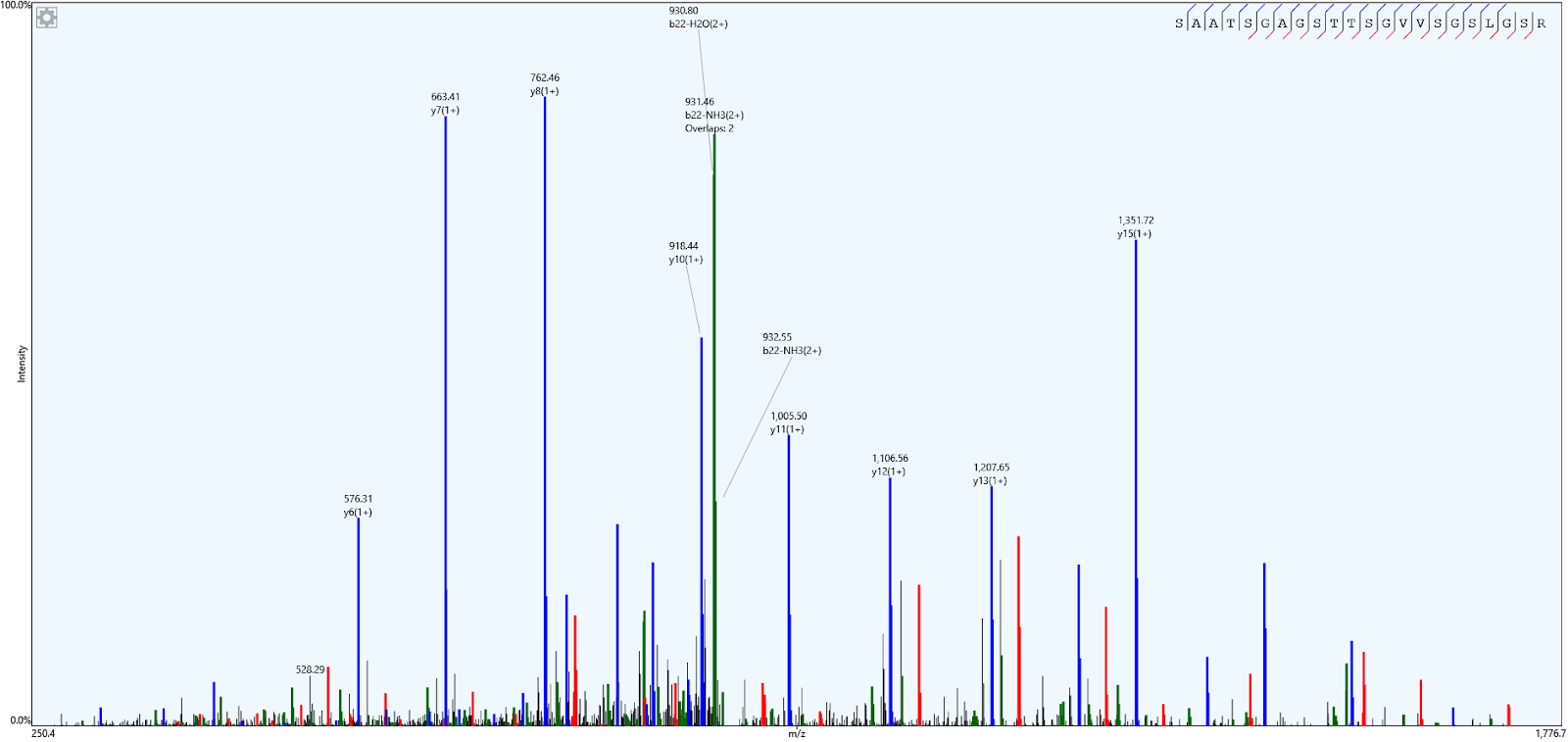 Figura 12 – Espectro de massas do peptídeo selecionado na amostra.
Figura 12 – Espectro de massas do peptídeo selecionado na amostra.
Além disso, a ferramenta oferece a funcionalidade de visualizar a cobertura da sequência, o que é fundamental para garantir que a proteína identificada está, de fato, presente na amostra analisada (Ver Figuras 13 e 14). Essa visualização da cobertura pode ser observada tanto em nível de sequência, mostrando quais regiões da proteína foram mapeadas pelos peptídeos, quanto em uma matriz de compartilhamento de peptídeos entre diferentes proteínas. Essa matriz fornece uma perspectiva adicional sobre as interações entre proteínas e a possibilidade de identificar peptídeos comuns, facilitando a compreensão das relações funcionais e das dinâmicas proteômicas dentro da amostra. Essa análise não apenas confirma a presença da proteína, mas também contribui para a interpretação biológica dos resultados, permitindo inferências mais robustas sobre os processos celulares em questão.
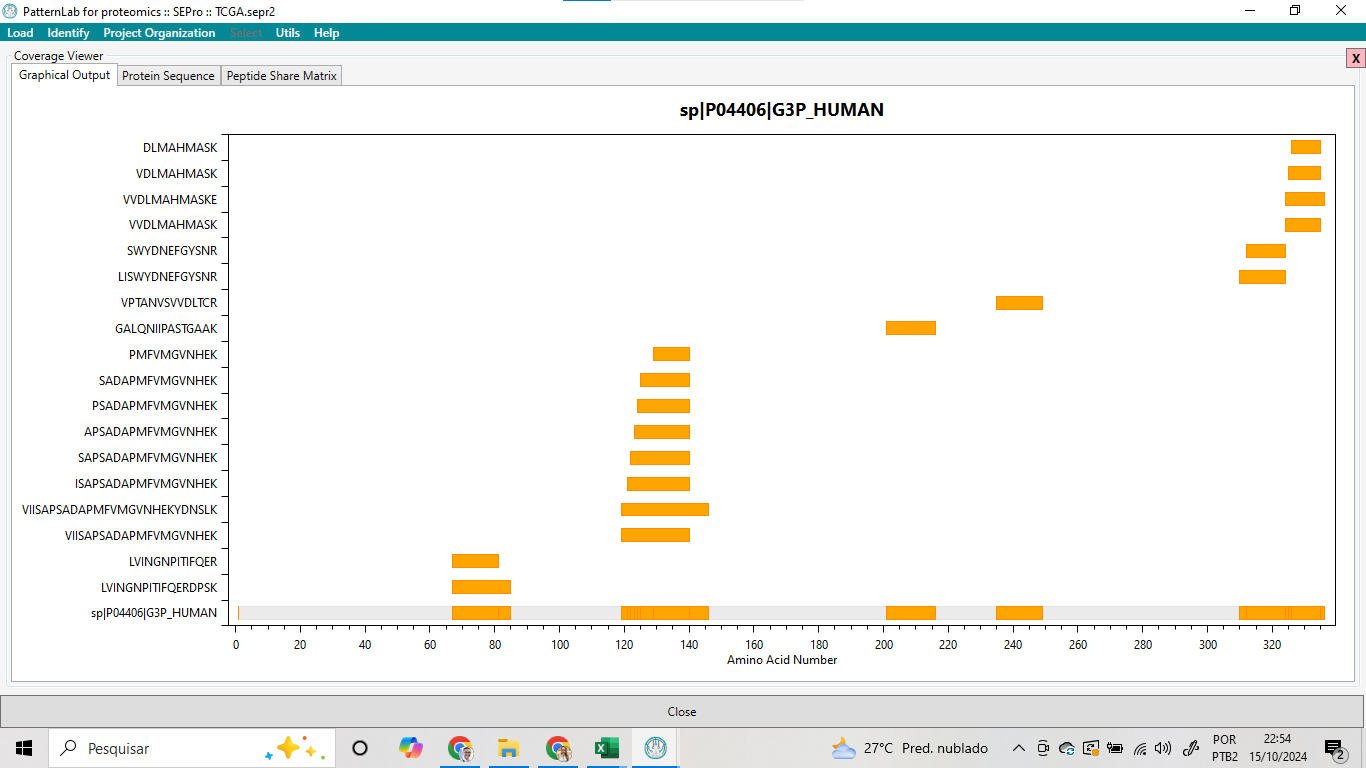 Figura 13 – A análise da cobertura proteica da proteína G3P_HUMAN (UniProt ID: P04406).
Figura 13 – A análise da cobertura proteica da proteína G3P_HUMAN (UniProt ID: P04406).
 Figura 14 – A análise da cobertura proteica da proteína G3P_HUMAN (UniProt ID: P04406) a nível de sequência.
Figura 14 – A análise da cobertura proteica da proteína G3P_HUMAN (UniProt ID: P04406) a nível de sequência.
A análise da cobertura proteica da proteína G3P_HUMAN (UniProt ID: P04406) revelou uma cobertura heterogênea ao longo da sequência de 335 aminoácidos (Figura 15). As regiões compreendidas entre os resíduos 120 e 150, assim como entre os resíduos 300 e 330, apresentaram uma maior cobertura, conforme indicado pelas barras laranjas no gráfico de cobertura. Nesses trechos, múltiplos peptídeos foram detectados, sugerindo que essas áreas da proteína são mais suscetíveis à digestão enzimática e/ou mais expostas durante o experimento. Em contrapartida, outras regiões, particularmente entre os resíduos 60-100 e 160-250, mostraram uma cobertura limitada ou ausente, o que pode indicar áreas de menor acessibilidade enzimática ou menor abundância nos fragmentos proteicos gerados.
Adicionalmente, a análise da Matriz de Compartilhamento de Peptídeos (Peptide Share Matrix) entre diferentes proteínas revelou que a proteína G3P_HUMAN compartilha 27 peptídeos com a proteína MC19_HUMAN (UniProt ID: Q9NX63), conforme destacado em amarelo (Figura 15). Essa alta sobreposição de peptídeos sugere uma similaridade significativa entre essas duas proteínas, o que pode estar relacionado a funções biológicas ou domínios estruturais conservados entre elas. Além disso, observou-se que a proteína AHNK_HUMAN (UniProt ID: Q09666) compartilha 9 peptídeos com a proteína MVP_HUMAN (UniProt ID: Q14764), enquanto TRIPC_HUMAN (UniProt ID: Q14669) compartilha 1 peptídeo com a mesma proteína.
Esses resultados indicam que há compartilhamento de peptídeos entre diferentes proteínas, o que pode ser útil para estudos de homologia e evolução proteica, além de fornecer insights sobre a redundância de peptídeos detectados durante a análise de espectrometria de massa.
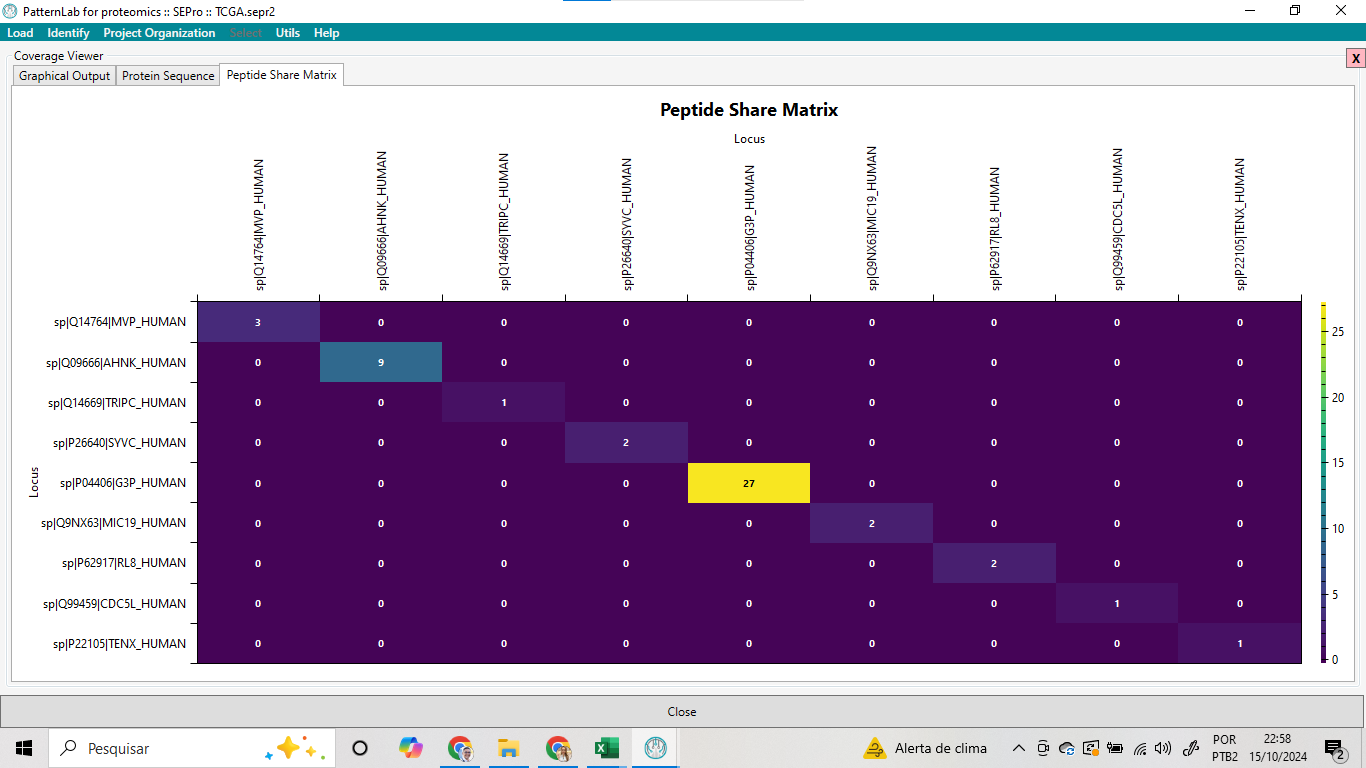 Figura 15 – Matriz de Compartilhamento de Peptídeos (Peptide Share Matrix)
Figura 15 – Matriz de Compartilhamento de Peptídeos (Peptide Share Matrix)
CONSIDERAÇÕES FINAIS
O PatternLab for Proteomics V é uma ferramenta robusta e eficaz para a análise proteômica, especialmente na detecção de variantes proteicas em estudos complexos, como o câncer. O software combina múltiplos módulos de análise em um ambiente integrado, facilitando a organização e interpretação de grandes volumes de dados de espectrometria de massas, técnica essencial para a análise proteômica.
Além de sua precisão na identificação de peptídeos variantes, que desempenham um papel fundamental no desenvolvimento de biomarcadores e alvos terapêuticos, o PatternLab for Proteomics V se destaca por ser uma ferramenta gratuita, desenvolvida pelo Laboratório de Proteômica Computacional e Estrutural da FIOCRUZ, o que o torna um produto nacional de alto impacto. Sua acessibilidade e integração de dados experimentais com bancos de dados de sequências proteicas conferem uma grande vantagem à pesquisa biomédica, fortalecendo a capacidade dos cientistas de avançarem no entendimento e tratamento de doenças complexas, como o câncer.
Devido ao seu caráter gratuito e à sua interface amigável, o PatternLab for Proteomics V é uma excelente opção para universidades públicas, especialmente aquelas que possuem menos recursos financeiros para investir em softwares caros de análise. O uso do PatternLab em instituições acadêmicas com recursos limitados pode aumentar a capacidade de realização de pesquisas de ponta, permitindo que grupos de pesquisa de diferentes níveis avancem na ciência de forma eficiente e sem custos adicionais, democratizando o acesso a tecnologias avançadas no campo da bioinformática e proteômica.
REFERÊNCIAS
CARVALHO, P.C. et al. PatternLab for proteomics: a tool for differential shotgun proteomics. BMC Bioinformatics 9, 316 (2008). https://doi.org/10.1186/1471-2105-9-316
CARVALHO, Paulo C. et al. YADA: a tool for taking the most out of high-resolution spectra. Bioinformatics, v. 25, n. 20, p. 2734-2736, 2009.
CARVALHO, Paulo C. et al. Search engine processor: filtering and organizing peptide spectrum matches. Proteomics, v. 12, n. 7, p. 944-949, 2012.
DA CUNHA, Lucas Marques et al. dbPepVar: a novel cancer proteogenomics database. IEEE Access, v. 10, p. 90982-90994, 2022.
DEN RIDDER, Maxime; DARAN-LAPUJADE, Pascale; PABST, Martin. Shot-gun proteomics: why thousands of unidentified signals matter. FEMS Yeast Research, v. 20, n. 1, p. foz088, 2020.
ENG, Jimmy K.; JAHAN, Tahmina A.; HOOPMANN, Michael R. Comet: an open‐source MS/MS sequence database search tool. Proteomics, v. 13, n. 1, p. 22-24, 2013.
FESENKO, Igor et al. Alternative splicing shapes transcriptome but not proteome diversity in Physcomitrella patens. Scientific reports, v. 7, n. 1, p. 2698, 2017.
GAO, Yu; YATES III, John R. Protein analysis by shotgun proteomics. Mass Spectrometry‐Based Chemical Proteomics, p. 1-38, 2019.
LI, Jing et al. A bioinformatics workflow for variant peptide detection in shotgun proteomics. Molecular & Cellular Proteomics, v. 10, n. 5, 2011.
LIN, Tai‐Tu et al. Mass spectrometry‐based targeted proteomics for analysis of protein mutations. Mass Spectrometry Reviews, v. 42, n. 2, p. 796-821, 2023.
CLASEN, Milan A. et al. PatternLab V handles multiplex spectra in shotgun proteomic searches and increases identification. Journal of the American Society for Mass Spectrometry, v. 34, n. 4, p. 794-796, 2023.
SANTOS, M.D.M. et al. Simple, efficient and thorough shotgun proteomic analysis with PatternLab V. Nat Protoc 17, 1553–1578 (2022). https://doi.org/10.1038/s41596-022-00690-x
SINITCYN, Pavel; RUDOLPH, Jan Daniel; COX, Jürgen. Computational methods for understanding mass spectrometry–based shotgun proteomics data. Annual Review of Biomedical Data Science, v. 1, n. 1, p. 207-234, 2018.
THANGUDU, Ratna Rajesh et al. Abstract LB-242: Proteomic Data Commons: A resource for proteogenomic analysis. Cancer Research, v. 80, n. 16_Supplement, p. LB-242-LB-242, 2020.