

A utilização de dados genômicos disponíveis em bancos públicos é uma estratégia eficiente para otimizar a validação de hipóteses, reduzindo custos e tempo. Este tutorial tem como objetivo fornecer um manual prático para utilizar dados públicos do Gene Expression Omnibus (GEO) na validação de experimentos de biologia molecular. Serão apresentados os passos necessários para acessar, processar e integrar esses dados com métodos experimentais tradicionais, como o quantitative Polymerase Chain Reaction (qPCR). O manual aborda o acesso ao GEO, como realizar buscas eficazes e a extração dos dados, além de instruções sobre o uso de pacotes do R, como limma e DESeq2, para o processamento e análise diferencial dos dados de expressão gênica. A seguir, é detalhado como os dados extraídos podem ser integrados com experimentos laboratoriais para validação dos resultados.
Autores:
Wanderson Gabriel Gomes de Melo (0000-0002-5120-2281)
Camile Benício Campêlo (0009-0001-3011-5779)
Regina Lucia dos Santos Silva (0000-0003-3773-2590)
Dayseanny de Oliveira Bezerra (0000-0002-7662-2776)Revisão: Izabela, Filipe Teixeira, Ana Carolina
Introdução
A biologia molecular tem desempenhado um papel fundamental no avanço das pesquisas em genômica e transcriptômica, especialmente com o advento de tecnologias como o RNA-seq [1,2]. Essa técnica, amplamente utilizada para mapear e quantificar transcritos, gera uma quantidade massiva de dados, cujos arquivos podem facilmente atingir terabytes de informação bruta [3]. No entanto, o processamento e a interpretação desses dados apresentam desafios consideráveis, exigindo recursos computacionais robustos e ferramentas bioinformáticas avançadas. A complexidade dos dados de RNA-seq envolve o grande volume de informações geradas, a necessidade de pipelines sofisticados para filtrar, mapear e quantificar a expressão gênica com precisão, e o alto custo associado ao processamento desses dados [4]. Esses fatores, combinados, frequentemente tornam a análise um gargalo em muitos projetos de pesquisa, resultando em atrasos na validação de hipóteses experimentais e na conclusão dos estudos [5].
Métodos tradicionais como o PCR quantitativo em tempo real (quantitative Polymerase Chain Reaction, qPCR ) e o western blot continuam sendo abordagens amplamente utilizadas na biologia molecular para a validação da expressão gênica e de proteínas, respectivamente [6,7]. Essas técnicas fornecem dados quantitativos e semi-quantitativos de forma mais acessível e rápida em comparação ao RNA-seq. Além disso, essas abordagens podem ser usadas de maneira complementar com bases de dados preexistentes, como as encontradas no Gene Expression Omnibus (GEO) e ArrayExpress, que abrigam uma vasta coleção de dados de expressão gênica de diversos organismos e condições experimentais [8]. O uso dessas bases de dados facilita a reanálise de dados e a integração de novos experimentos, permitindo a validação de resultados experimentais em um contexto mais amplo.
O objetivo deste artigo é apresentar um tutorial que guie o leitor em como utilizar dados disponíveis em bancos públicos, como o GEO, para validar experimentos de biologia molecular, demonstrando como acessar, processar e integrar esses dados em conjunto com resultados experimentais obtidos por técnicas tradicionais, otimizando a validação de hipóteses e a análise dos resultados.
Primeira parte – data minning
Nesta primeira parte do tutorial, serão apresentados os passos necessários para realizar a extração de dados de expressão gênica de bancos públicos utilizando o GEO. O objetivo é encontrar datasets (conjuntos de dados organizados e estruturados) relevantes que possam ser usados para validar seus experimentos de biologia molecular, o organograma completo encontra-se em figura 1.
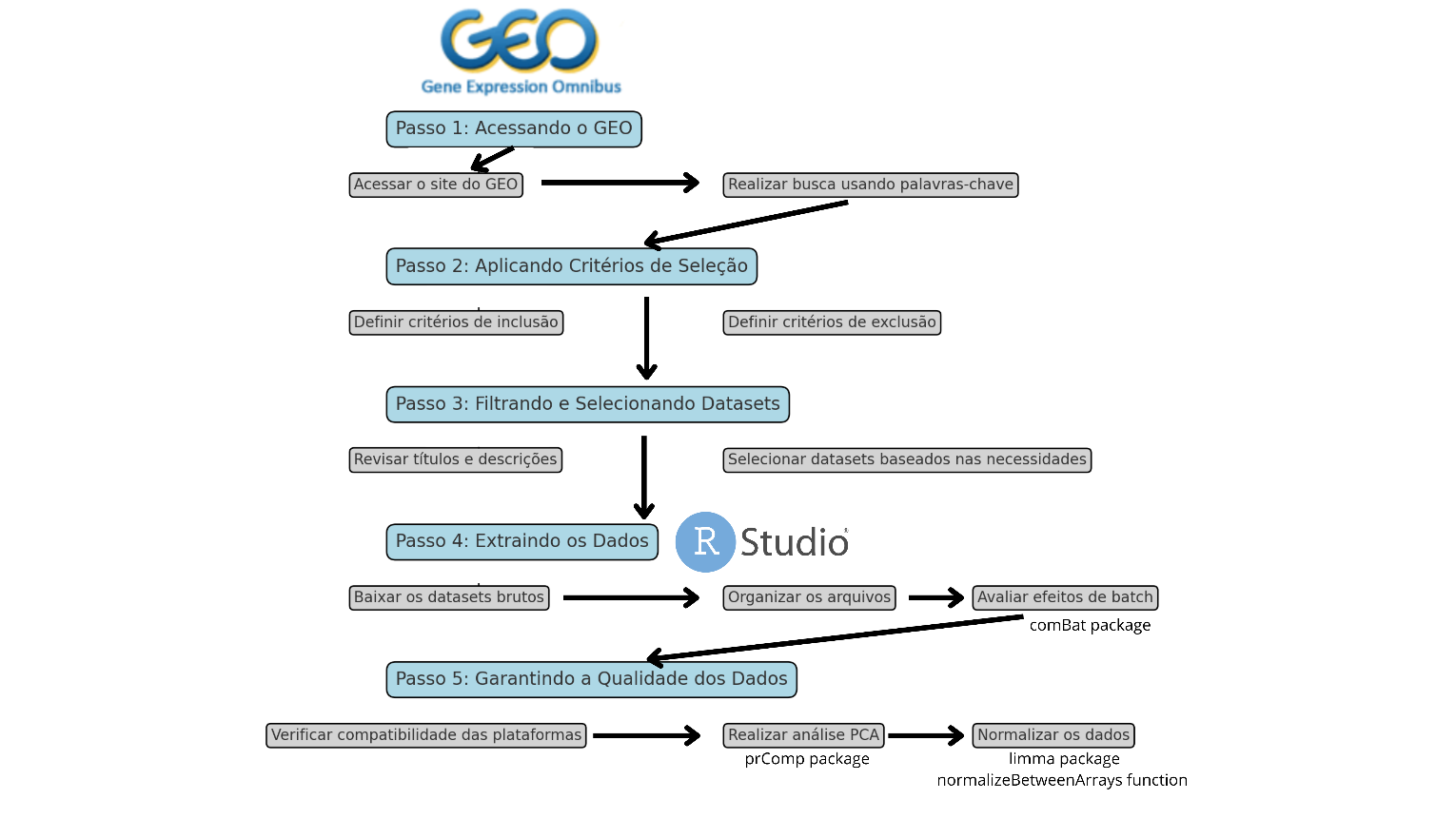 Figura 1 – Organograma contendo passo-a-passo para mineração e aquisição de dados nos arquivos indexados no GEO.
Figura 1 – Organograma contendo passo-a-passo para mineração e aquisição de dados nos arquivos indexados no GEO.
Passo 1 – Acessando o GEO
-
Acesse o site do GEO: O GEO é um banco de dados público hospedado pelo NCBI e pode ser acessado diretamente através do link: https://www.ncbi.nlm.nih.gov/geo/ [9].
-
Realize a busca por datasets: Na página inicial do GEO, você verá uma barra de busca. Utilize palavras-chave relacionadas ao seu experimento, como “macrophages”, “RNA-seq”, ou termos específicos sobre as células ou condições experimentais de interesse. Busque por palavras-chave relevantes ao tema da sua pesquisa, utilizando MESH terms e operadores booleanos, assim como é feito em buscas no PubMed (Figura 2).
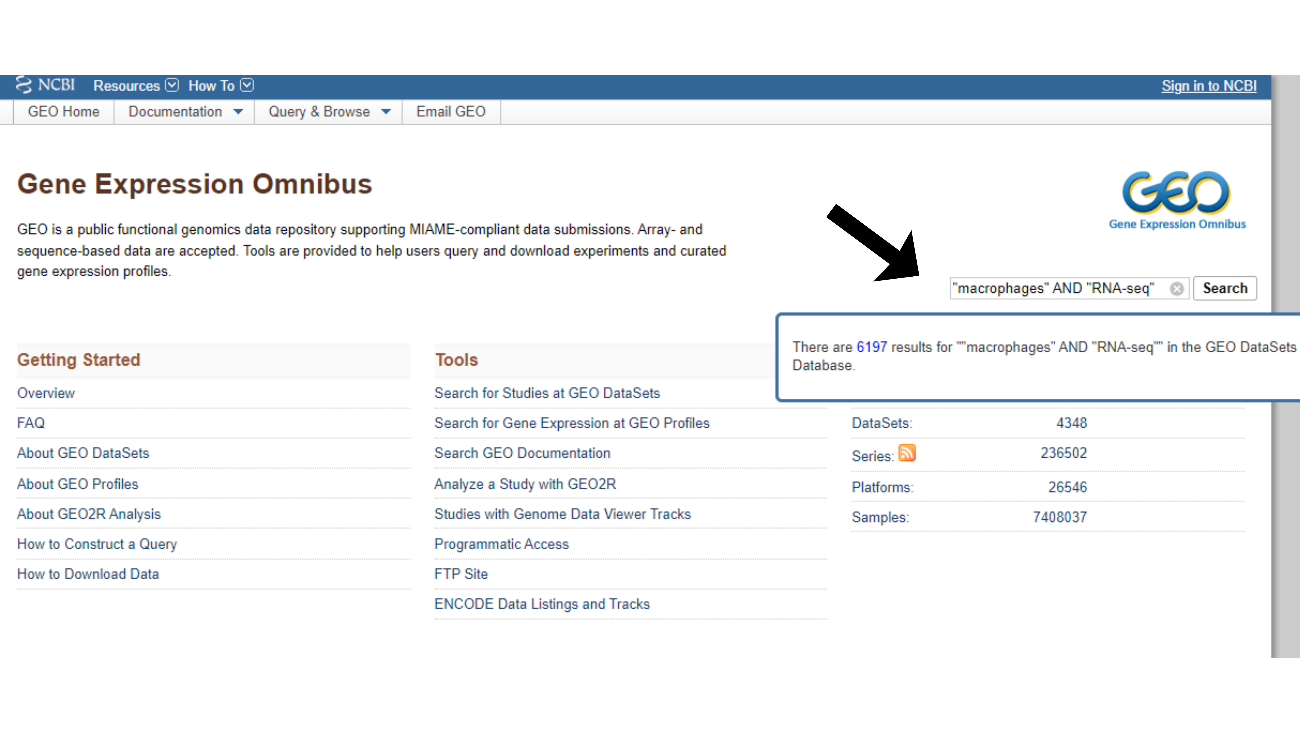 Figura 2 – Tela de busca de datasets nas bases de dados do GEO utilizando palavras-chave no padrão MESH com operadores booleanos (seta preta).
Figura 2 – Tela de busca de datasets nas bases de dados do GEO utilizando palavras-chave no padrão MESH com operadores booleanos (seta preta).
Passo 2 – Aplicando Critérios de Seleção
-
Defina os critérios de inclusão: Para refinar sua busca, aplique critérios de inclusão, como:
-
Tipo de tecnologia utilizada para aquisição de dados (o tipo de tecnologia irá definir o grau de dificuldade do processamento dos dados),
-
Período de tempo específico,
-
Espécie,
-
Tipo de célula ou órgão específico,
-
Tratamento específico,
-
Número de amostras e/ou tratamentos,
-
Replicatas técnicas e biológicas.
-
-
Defina critérios de exclusão: exclua datasets irrelevantes – remova estudos que não atendem ao seu objetivo experimental.
-
A figura 3 mostra critérios de seleção usados na busca realizada no item anterior.
 Figura 3 – Critérios de inclusão e exclusão de dados utilizados na busca de datasets no GEO.
Figura 3 – Critérios de inclusão e exclusão de dados utilizados na busca de datasets no GEO.
Passo 3: Filtrando e Selecionando Datasets
-
Analise os resultados: Após refinar sua busca, será obtida uma lista de estudos relevantes. Revise os títulos e descrições para verificar se correspondem às suas necessidades experimentais,
-
Selecione os datasets mais apropriados: Ao final da análise, escolha os estudos que melhor atendem aos seus critérios,
-
Após selecionar os datasets mais apropriados para seu experimento, é essencial garantir que os dados escolhidos sejam comparáveis e confiáveis. Nesta etapa, abordaremos alguns fatores críticos para assegurar a qualidade e a validade da análise:
-
Verifique a compatibilidade das plataformas utilizadas: Certifique-se de que os datasets selecionados foram gerados em plataformas tecnológicas compatíveis, como RNA-seq ou microarray, e que utilizam a mesma tecnologia de detecção (e.g., Illumina HiSeq ou Affymetrix). A equivalência de plataformas é crucial para garantir que as diferenças observadas nos dados sejam atribuídas às variáveis experimentais, e não a possíveis variações técnicas entre plataformas,
-
Evite comparar dados de diferentes plataformas diretamente: A integração de dados de diferentes tecnologias requer métodos de harmonização avançados, que podem introduzir erros. Sempre que possível, prefira dados gerados pela mesma plataforma,
-
Figuras 4 e 5 mostram datasets com destaque nas plataformas utilizadas para obtenção de dados.
-
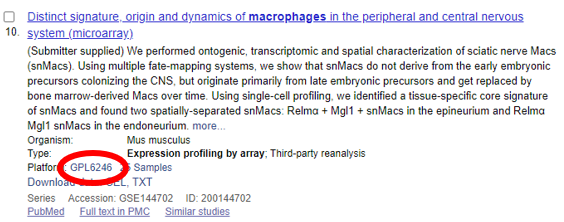 Figura 4 – Dataset do banco de dados GEO número GSE144702 contendo dados de expressão gênica por array avaliados por meio da plataforma GPL6246 (Affymetrix Mouse Gene 1.0 ST Array) (círculo vermelho) [10].
Figura 4 – Dataset do banco de dados GEO número GSE144702 contendo dados de expressão gênica por array avaliados por meio da plataforma GPL6246 (Affymetrix Mouse Gene 1.0 ST Array) (círculo vermelho) [10].
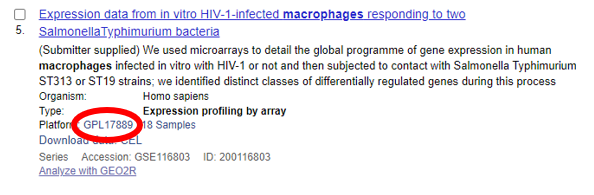 Figura 5 – Dataset do banco de dados GEO número GSE116803 contendo dados de expressão gênica por array avaliados por meio da plataforma GPL17889 (Affymetrix Human Gene 2.0 ST Array) (círculo vermelho) [11].
Figura 5 – Dataset do banco de dados GEO número GSE116803 contendo dados de expressão gênica por array avaliados por meio da plataforma GPL17889 (Affymetrix Human Gene 2.0 ST Array) (círculo vermelho) [11].
Segunda parte – Data extraction
Passo 4 – Baixe os datasets selecionados
Clique no título do dataset escolhido e vá até a seção de “Downloads”. Baixe os arquivos de dados brutos (.CEL, .TXT, ou outro formato), bem como os metadados associados (informações sobre as amostras, plataformas usadas, e condições experimentais) (Figura 6).
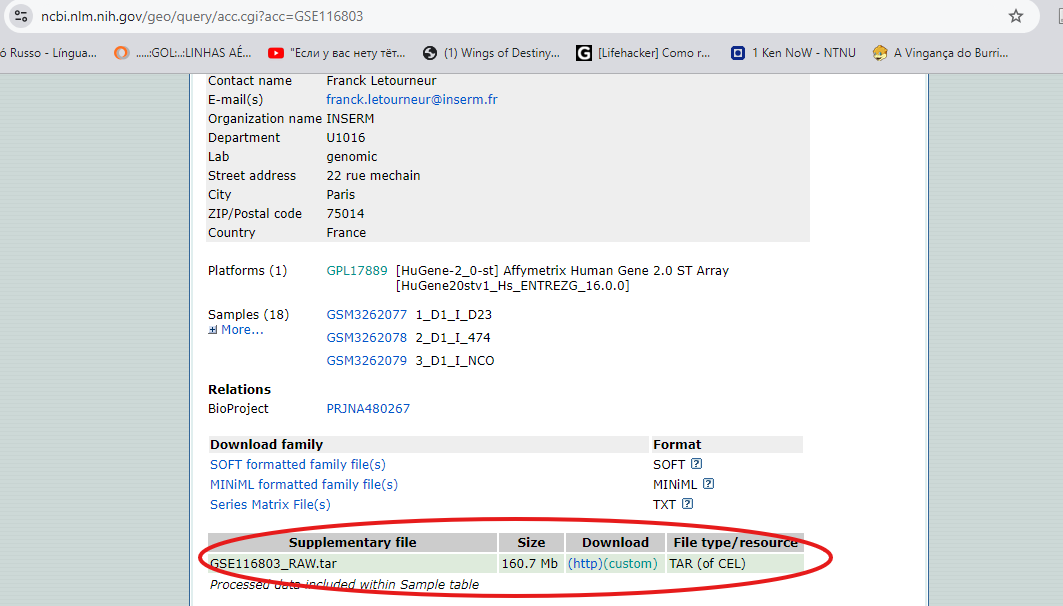 Figura 6 – Protocolo para realização de download de dados em formato “.CEL” (círculo vermelho), para posterior análise.
Figura 6 – Protocolo para realização de download de dados em formato “.CEL” (círculo vermelho), para posterior análise.
Passo 5 – Prepare os arquivos para análise
Após o download, organize os arquivos em pastas, separando os dados brutos e os metadados para facilitar o processamento nos próximos passos.
Passo 6 – Identifique e avalie possíveis efeitos de batch
Muitos estudos contêm lotes (batches) experimentais diferentes, que podem introduzir variações indesejadas nos dados. Essas variações podem mascarar as diferenças biológicas reais. Ao revisar os datasets, verifique se os lotes estão claramente identificados e considere métodos de correção de batch, como o uso da ferramenta Combat no R, para eliminar essas variações [12].
Passo 7 – Realize uma análise Principal Component Analysis (PCA) para identificar variâncias nos dados:
A PCA é uma ferramenta poderosa que permite visualizar a distribuição dos dados e identificar agrupamentos naturais, bem como detectar amostras outliers ou influências indesejadas de batch. Após integrar os datasets, a realização de uma PCA ajuda a confirmar se os grupos experimentais estão adequadamente separados, o que pode indicar uma boa qualidade dos dados [13].
Passo 8 – Normalização é essencial para garantir a comparabilidade
Os datasets de expressão gênica, especialmente quando oriundos de diferentes estudos, precisam ser normalizados antes da análise final. Utilize pacotes como DESeq2 ou limma para normalizar os dados de RNA-seq ou microarray, respectivamente [14–16]. A normalização corrige variações técnicas entre amostras e ajusta os dados para que as comparações sejam feitas de forma precisa, refletindo diferenças biológicas reais.
A equivalência de plataformas, a correção de batch effects, a análise de PCA e a normalização adequada são etapas críticas para garantir que os dados selecionados possam ser comparados de maneira robusta e válida. Ignorar essas etapas pode comprometer a integridade dos resultados e levar a conclusões errôneas. Ao seguir essas práticas, você assegura a qualidade e a confiabilidade das análises subsequentes.
Parte 3 – Analisando os Dados
Passo 9 – Análise em software
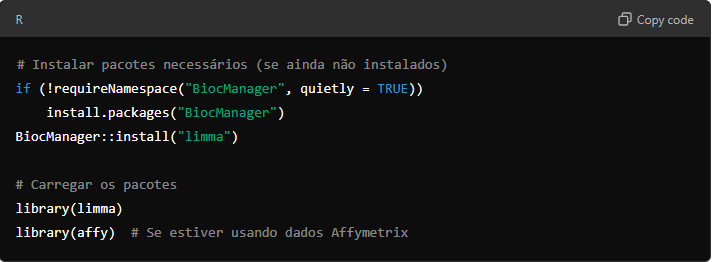
Depois de baixar, filtrar e normalizar os dados dos datasets selecionados, a próxima etapa é realizar a análise diferencial de expressão gênica. Neste tutorial, utilizaremos o pacote limma no R para analisar dados de microarray. O limma (Linear Models for Microarray Data) é amplamente utilizado para análise de dados de microarrays e RNA-seq, permitindo a identificação de genes diferencialmente expressos (DEGs) entre grupos experimentais.
9.1. Carregando os Dados
Primeiro, certifique-se de que seus arquivos de dados brutos (.CEL ou .TXT) e metadados estão prontos para serem carregados no R.
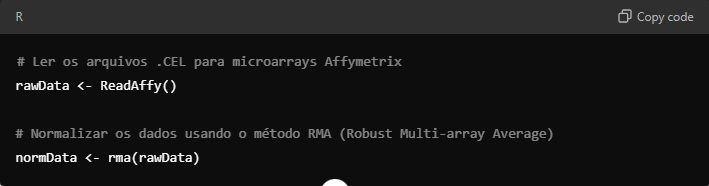
Em seguida, importe os dados brutos:
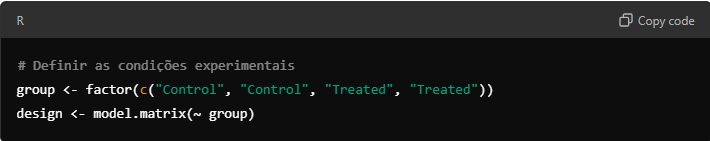
9.2. Preparando o Design Experimental
Você precisa criar uma matriz de design que descreva as condições experimentais dos seus dados (por exemplo, controle vs. tratado). Isso permite ao limma ajustar os modelos lineares.
9.3. Ajustando o Modelo Linear
Após definir o design experimental, ajuste o modelo linear aos dados de expressão.

Aplicando Contrastes para Comparar Grupos – agora, defina os contrastes que você deseja comparar. Por exemplo, comparar “Control” vs “Treated”.
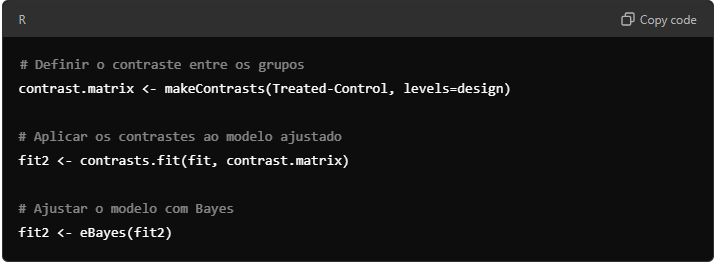
9.4. Identificando Genes Diferencialmente Expressos
Use a função topTable para identificar os genes diferencialmente expressos com base em um valor de corte de significância (por exemplo, p < 0,05).
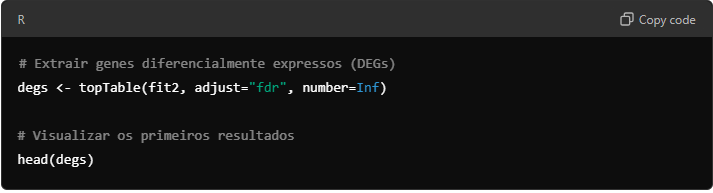
9.5. Gerando um Volcano Plot
Para visualizar os resultados da análise, você pode criar um Volcano Plot, que destaca os genes com maior significância estatística e alteração de expressão (log-fold change).
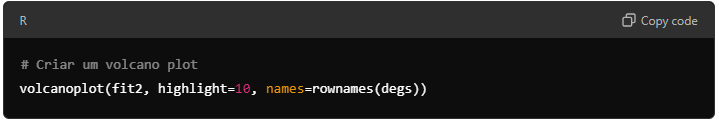
9.6. Análise geral
Os genes que aparecem na tabela de resultados (DEGs) serão aqueles que passaram pelo filtro de significância ajustado pelo limma (eBayes). Esses genes podem então ser analisados em maior detalhe ou usados para validação experimental em qPCR, por exemplo.
Conclusão
Este tutorial demonstra como utilizar dados públicos disponíveis no Gene Expression Omnibus (GEO) para validar experimentos de biologia molecular, integrando essas informações com técnicas experimentais tradicionais, como qPCR. A utilização de dados genômicos de fontes públicas é uma abordagem poderosa para otimizar a validação de hipóteses, permitindo a redução de custos e o aproveitamento de dados previamente gerados. O uso de ferramentas como limma e DESeq2 proporciona uma análise robusta dos dados de expressão gênica, identificando genes diferencialmente expressos de forma eficiente e confiável. Além disso, esses dados podem atuar como um screening inicial para definir genes-alvo de uma pesquisa ou servir como critério para a validação experimental, ampliando o contexto dos resultados obtidos em laboratório.
Ao incorporar dados públicos e métodos computacionais avançados, os pesquisadores podem acelerar suas descobertas, validar suas hipóteses com maior precisão e gerar resultados que estejam em sintonia com os achados globais na área de biologia molecular e transcriptômica. Assim, o uso dessas bases de dados se revela uma ferramenta indispensável na era da ciência de dados e na biologia molecular moderna.
Referências
[1] M.H.H. Withanage, H. Liang, E. Zeng, RNA-Seq Experiment and Data Analysis, Methods Mol Biol 2418 (2022) 405–424. https://doi.org/10.1007/978-1-0716-1920-9_22.
[2] H. Miura, R.T. Cerbus, I. Noda, I. Hiratani, CWL-Based Analysis Pipeline for Hi-C Data: From FASTQ Files to Matrices, Methods Mol Biol 2856 (2025) 79–117. https://doi.org/10.1007/978-1-0716-4136-1_6.
[3] U. Singh, J. Li, A. Seetharam, E.S. Wurtele, pyrpipe: a Python package for RNA-Seq workflows, NAR Genom Bioinform 3 (2021) lqab049. https://doi.org/10.1093/nargab/lqab049.
[4] M.D. Luecken, F.J. Theis, Current best practices in single-cell RNA-seq analysis: a tutorial, Mol Syst Biol 15 (2019) e8746. https://doi.org/10.15252/msb.20188746.
[5] C.S. Pareek, R. Smoczynski, A. Tretyn, Sequencing technologies and genome sequencing, J Appl Genet 52 (2011) 413–435. https://doi.org/10.1007/s13353-011-0057-x.
[6] F. Li, W. Si, L. Xia, D. Yin, T. Wei, M. Tao, X. Cui, J. Yang, T. Hong, R. Wei, Positive feedback regulation between glycolysis and histone lactylation drives oncogenesis in pancreatic ductal adenocarcinoma, Mol Cancer 23 (2024) 90. https://doi.org/10.1186/s12943-024-02008-9.
[7] Y. Wu, J. Wang, T. Zhao, J. Chen, L. Kang, Y. Wei, L. Han, L. Shen, C. Long, S. Wu, G. Wei, Di-(2-ethylhexyl) phthalate exposure leads to ferroptosis via the HIF-1α/HO-1 signaling pathway in mouse testes, J Hazard Mater 426 (2022) 127807. https://doi.org/10.1016/j.jhazmat.2021.127807.
[8] H. Jiang, X. Zhang, Y. Wu, B. Zhang, J. Wei, J. Li, Y. Huang, L. Chen, X. He, Bioinformatics identification and validation of biomarkers and infiltrating immune cells in endometriosis, Front Immunol 13 (2022) 944683. https://doi.org/10.3389/fimmu.2022.944683.
[9] Home – GEO – NCBI, (n.d.). https://www.ncbi.nlm.nih.gov/geo/ (accessed September 22, 2024).
[10] E. Ydens, L. Amann, B. Asselbergh, C.L. Scott, L. Martens, D. Sichien, O. Mossad, T. Blank, S. De Prijck, D. Low, T. Masuda, Y. Saeys, V. Timmerman, R. Stumm, F. Ginhoux, M. Prinz, S. Janssens, M. Guilliams, Profiling peripheral nerve macrophages reveals two macrophage subsets with distinct localization, transcriptome and response to injury, Nat Neurosci 23 (2020) 676–689. https://doi.org/10.1038/s41593-020-0618-6.
[11] Expression data from in vitro HIV-1-infected macrophages responding to two SalmonellaTyphimurium bacteria – – GEO DataSets – NCBI, (n.d.). https://www.ncbi.nlm.nih.gov/gds/?term=GSE116803[Accession] (accessed September 22, 2024).
[12] Y. Zhang, G. Parmigiani, W.E. Johnson, ComBat-seq: batch effect adjustment for RNA-seq count data, NAR Genom Bioinform 2 (2020) lqaa078. https://doi.org/10.1093/nargab/lqaa078.
[13] H. Lee, B. Han, FastRNA: An efficient solution for PCA of single-cell RNA-sequencing data based on a batch-accounting count model, Am J Hum Genet 109 (2022) 1974–1985. https://doi.org/10.1016/j.ajhg.2022.09.008.
[14] M.E. Ritchie, B. Phipson, D. Wu, Y. Hu, C.W. Law, W. Shi, G.K. Smyth, limma powers differential expression analyses for RNA-sequencing and microarray studies, Nucleic Acids Res 43 (2015) e47. https://doi.org/10.1093/nar/gkv007.
[15] S. Liu, Z. Wang, R. Zhu, F. Wang, Y. Cheng, Y. Liu, Three Differential Expression Analysis Methods for RNA Sequencing: limma, EdgeR, DESeq2, J Vis Exp (2021). https://doi.org/10.3791/62528.
[16] M.I. Love, W. Huber, S. Anders, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, Genome Biol 15 (2014) 550. https://doi.org/10.1186/s13059-014-0550-8.