

Revisão:
BIOINFO – Revista Brasileira de Bioinformática. Edição #. .
DOI:
Cobertura é um conceito bastante citado quando se trata de sequenciamento de genomas. Dependendo do contexto, o conceito de cobertura pode ter diferentes significados, o que pode gerar um pouco de confusão. Neste artigo, serão apresentadas terminologias para diferenciar os dois tipos de cobertura: vertical e horizontal. Em sequenciamento NGS, cobertura vertical indica a quantidade média de leituras que cobrem cada região de um genoma com base no mapeamento (por exemplo, profundidade de cobertura de 30x). Cobertura horizontal indica o percentual estimado do genoma que foi sequenciado, ou seja, o quanto do genoma final está representado no total de bases sequenciadas (por exemplo, 95% de amplitude de cobertura).
Introdução
Durante o projeto de sequenciamento do genoma humano, o conceito de cobertura era utilizado para identificar o quanto faltava para conclusão. Temos que levar em consideração que o Projeto Genoma Humano levou 13 anos para ser concluído. Logo, naquela época, era relevante saber, à medida do tempo, qual o percentual dos três bilhões de pares de base (3 Gb) do genoma humano já havia sido identificado e quanto ainda faltava. Com o surgimento da técnica de sequenciamento de Shotgun, o conceito de cobertura ganhou um novo sentido.
O sequenciamento Shotgun, também denominado como sequenciamento WGS (Whole Genome Shotgun), requer etapas de fragmentação e amplificação (Figura 1). Essas etapas são necessárias uma vez que os sequenciadores de alta performance conseguiam identificar apenas pequenos fragmentos de DNA. Leve em consideração que um genoma possui aproximadamente 3.000.000.000 pb e um sequenciador da época só conseguia identificar fragmentos bem menores do que 1.000 pb. Apesar disso, a estratégia de Shotgun representou uma revolução, uma vez que permitia que uma imensa quantidade de fragmentos fosse lida ao mesmo tempo, reduzindo consideravelmente o tempo necessário para o sequenciamento. Cabia depois a computadores identificar a ordem correta dos fragmentos, estabelecendo assim a sequência completa do genoma (popularmente conhecido como o processo de montagem do genoma).
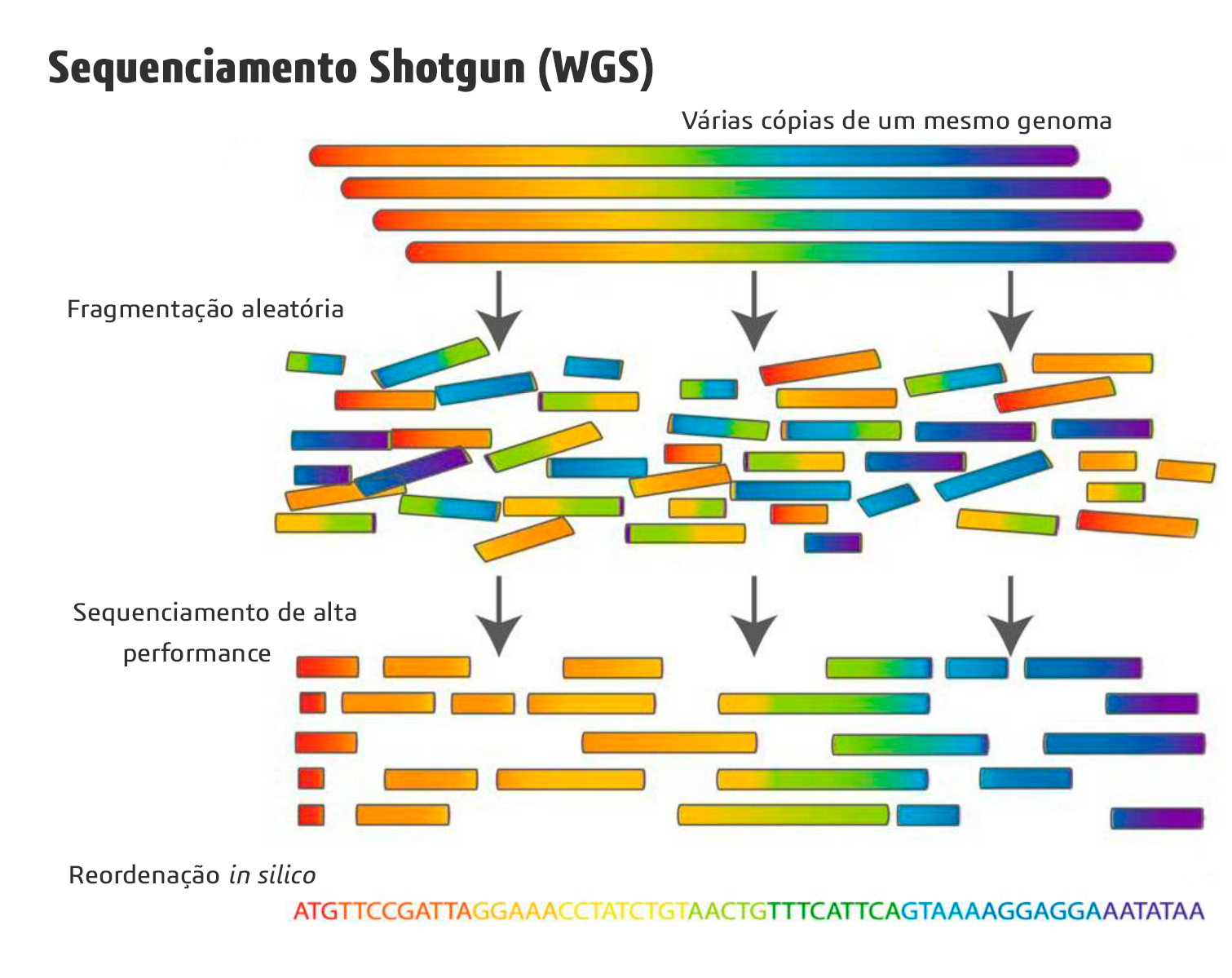
Entretanto, nem tudo são flores! Na teoria, várias cópias do genoma completo fragmentadas em posições aleatórias poderiam ser reordenadas por meio de algoritmos de montagem. Mas na prática, diversos problemas dificultam esse processo. Algumas regiões do genoma podem ser perdidas durante o processo de fragmentação e/ou amplificação. Outras podem ser menos propensas ao tipo de sequenciamento adotado (vide o caso dos homopolímeros na plataforma Ion Torrent, em que há grande dificuldade em identificar repetições de um mesmo nucleotídeo em sequência, como a sequência “TTTTTT”). Além disso, em determinados genomas há sequências repetitivas que atrapalham o processo de reordenação in silico, como regiões de transposons, repetições tandem e regiões codificadoras de RNA ribossomal. Com tudo isso, é possível concluir que, ao ordenar todos os fragmentos lidos em um sequenciamento, nem todas as partes do genoma serão representadas por uma mesma quantidade de sequências.
Falando de uma forma bem leiga, imagine que desejamos sequenciar um pequeno fragmento hipotético de DNA representado no quadro vermelho (Figura 2). Finja que não conhecemos a sequência final. Então, seis cópias dessa mesma sequência foram cortadas em fragmentos de 10 a 20 nucleotídeos e, em seguida, sequenciadas (vamos simplificar alguns detalhes do processo para facilitar o entendimento). Tenha em mente que apenas as sequências no quadro cinza (Figura 2) são conhecidas após o sequenciamento. Devemos ordená-las para obter a sequência original (também chamada de sequência consenso). Ao sobrepô-las, deveríamos ser capazes de obter seis cópias exatas da sequência final, mas perceba que isso não acontece. Tome como referência, por exemplo, o 27º nucleotídeo da sequência (o “A” grifado em preto na Figura 2). Veja que, na caixa cinza, apenas três deles estão empilhados. Nesse caso, dizemos que o 27º par de base da sequência possui uma cobertura de 3x. Veja que outros trechos possuem uma cobertura maior (como o G na posição 29) e outros menor (como o A na primeira posição). A cobertura final será a média do número de vezes em que cada base é representada nas leituras (ou a soma do total de nucleotídeos nas leituras dividido pelo tamanho final da sequência). No exemplo abaixo, a sequência tem 67 pb e as leituras têm, somadas, 160 pb. Logo, temos uma cobertura média de aproximadamente 2,4x.

Agora, digamos que um experimento de bancada indicou que o fragmento de DNA deveria ter 100 pares de base. Entretanto, o processo de montagem indicou que apenas 67 pares de base foram identificados. Trinta e três pares de base foram perdidos em algum momento. Dessa forma, não aparecem na nossa montagem final. Isso indica que conseguimos alcançar apenas 67% da cobertura esperada.
Definição de cobertura
No exemplo anterior, dissemos que a sequência possuía uma cobertura de 2,4x e de 67%. Qual o valor indica a real cobertura? A resposta é ambos. Podemos dizer que 2,4x é a profundidade de cobertura e 67% é a largura da cobertura no genoma final.
Em um trabalho em 2014, Sims e colaboradores [3] definiram que a amplitude de cobertura (breadth of coverage) de um genoma é o percentual de bases do genoma original que puderam ser identificadas. Na teoria, espera-se que 100% das bases sejam representadas em um bom sequenciamento, ou seja, cobertura de 100%. Já o número de vezes que uma base específica é representada é denominado como profundidade da cobertura (depth of coverage) [3].
Em minha dissertação de mestrado [2], optei por diferenciar os dois tipos de cobertura como largura de cobertura, para representar o percentual de bases sequenciadas, e profundidade de cobertura, para indicar o valor médio que cada base é representada. Entretanto, em uma conversa informal que tive alguns anos atrás, o Prof. Dr. Henrique Figueiredo do departamento de veterinária da Universidade Federal de Minas Gerais propôs uma forma mais facilmente compreensíveis de se diferenciar os tipos de cobertura, separando-as em horizontal e vertical. A cobertura horizontal seria o percentual do genoma representado nas leituras (equivalente à largura de cobertura) e a cobertura vertical seria a quantidade média de vezes que cada base é representada (ou seja, a profundidade de cobertura). A Figura 3 ilustra os dois tipos de cobertura: vertical e horizontal.

Glossário de sinônimos
Cobertura horizontal: cobertura, amplitude de cobertura, largura de cobertura, percentual do genoma sequenciado.
Cobertura vertical: cobertura, profundidade de cobertura, cobertura esperada (calculada antes do sequenciamento), cobertura empírica por base (calculada pós-sequenciamento).
Referências
[1] Commins, J., Toft, C., Fares, M. A. – “Computational Biology Methods and Their Application to the Comparative Genomics of Endocellular Symbiotic Bacteria of Insects.” Biol. Procedures Online (2009).
[2] Mariano, D. SIMBA: uma ferramenta Web para gerenciamento de montagens de genomas bacterianos. Dissertação de mestrado. Universidade Federal de Minas Gerais (2015).
[3] SIMS, David; SUDBERY, Ian; ILOTT, Nicholas E.; HEGER, Andreas; PONTING, Chris P. Sequencing depth and coverage: key considerations in genomic analyses. Nature Reviews. Volume 15, p. 121-132. 2014.